Practice Essentials
Ehlers-Danlos syndrome (EDS) consists of a group of inherited heterogeneous disorders that share a common decrease in the tensile strength and integrity of the skin, joints, and other connective tissues. [1] This group of connective-tissue disorders is characterized by abnormal collagen synthesis causing hyperextensibility of the skin, hypermobility of the joints, [2] and tissue fragility, as is seen by easy bruising and delayed wound healing with atrophic scarring. [3] Depending on the type, EDS can be diagnosed through laboratory studies or clinical examination. Once the syndrome has been diagnosed, preventative measures should be taken.
People with lax joints and multiple scars were first described in the medical writings of Hippocrates, dating back to 400 BCE. [4] In 1892, Dr. A. Tschernogobow, a Russian dermatologist, presented two case studies of patients who had marked loose, fragile skin and hypermobile large joints, to the Moscow Venereology and Dermatology Society. His work provides the first detailed clinical description of EDS. [3, 5]
The syndrome derives its name from additional clinical case reports presented by two physicians: Edvard Ehlers, a Danish dermatologist, in 1901, and Henri-Alexandre Danlos, a French physician with expertise in chemistry of skin disorders, in 1908. Both physicians combined the pertinent features of the syndrome and accurately delineated the phenotypic features of this group of inherited disorders. The name, Ehlers-Danlos syndrome, was coined in 1936. [3]
Some patients with EDS can demonstrate amazing, almost unnatural, contortions, often arousing the curiosity of onlookers. Niccolo Paganini (1782-1840) the famous Italian violinist, who was capable of miraculous feats in his playing owing to his hypermobile and loose joints, had phenotypic traits of EDS. [6] In the late 19th century, historians described performers with traits of EDS who displayed their hyperextensible maneuvers publicly in circuses and travelling shows. Some achieved celebrity status, acquiring titles such as "The India Rubber Man," "The Elastic Lady," and "The Human Pretzel."
Patients displaying clinical capabilities such as these raise suspicion of the diagnosis when identified on physical examination. Unfortunately, patients with EDS are often not diagnosed for many years. [7] Examples of hyperextensibility and hypermobility are shown in the following images.
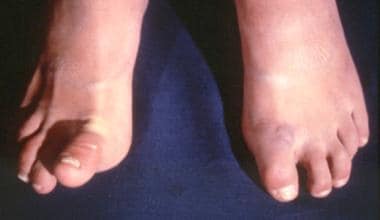 Patient with Ehlers-Danlos syndrome. Note the abnormal ability to elevate the right toe. Courtesy of Enrico Ceccolini, MD.
Patient with Ehlers-Danlos syndrome. Note the abnormal ability to elevate the right toe. Courtesy of Enrico Ceccolini, MD.
 Girl with Ehlers-Danlos syndrome. Dorsiflexion of all the fingers is easy and absolutely painless. Courtesy of Enrico Ceccolini, MD.
Girl with Ehlers-Danlos syndrome. Dorsiflexion of all the fingers is easy and absolutely painless. Courtesy of Enrico Ceccolini, MD.
 Patient with Ehlers-Danlos syndrome mitis. Joint hypermobility is less intense than with other conditions. Courtesy of Enrico Ceccolini, MD.
Patient with Ehlers-Danlos syndrome mitis. Joint hypermobility is less intense than with other conditions. Courtesy of Enrico Ceccolini, MD.
Signs and symptoms of Ehlers-Danlos syndrome
All EDS types share the following cutaneous features:
-
Loose, hyperextensible, fragile skin
-
Poor wound healing
-
Tendency to bruise easily
-
Atrophic scars that are wide and thin, known as "cigarette paper scars"
The following extracutaneous features are also shared:
-
Fragility of blood vessels
-
Hyperextensible, hypermobile joints
-
Propensity for spontaneous joint dislocations/subluxations
Workup in Ehlers-Danlos syndrome
The following laboratory studies may be indicated in patients diagnosed with EDS:
-
Diagnosis of the vascular-type EDS (type IV), arthrochalasia-type EDS (types VIIA and VIIB), and dermatosparaxis-type EDS (type VIIC) requires a skin biopsy; biochemical studies performed on cultured skin fibroblasts can detect alterations in collagen molecules
-
Molecular (DNA-based) testing is available for the vascular, arthrochalasia, and dermatosparaxis types
-
Kyphoscoliosis-type EDS (type VI) can be identified by urine enzyme assay
The remaining types of EDS—classical-type EDS and hypermobility-type EDS—are diagnosed through clinical examination. [8]
Management
Recommend low-resistance exercise for patients with EDS to help increase muscle tone and stabilize loose joints. Physical therapy guided by a therapist who is experienced in working with patients with connective-tissue abnormalities and joint dysfunction can be very helpful in the management of long-term health.
Instruct patients with EDS to avoid excessive or repetitive heavy lifting and other movements that produce undue strain or stress on their already hypermobile joints (eg, finger hyperextension that occurs with pushing off of a wall with the palms of the hands). [9] Advise patients to minimize joint trauma by avoiding joint hyperextension or joint locking.
Surgery may be indicated to correct fractures and stabilize dislocated joints. Preferential use of staples or tape (rather than stitches) for wound closure should be strongly considered. [10]
Comprehensive, accurate genetic counseling is one of the most critical issues in the treatment of patients with EDS. Provide the family with detailed information regarding EDS’s inheritance pattern and recurrence risks, as well as identification of at-risk family members. Screen pertinent individuals in the family for subtle signs and symptoms of the condition, regardless of whether signs or symptoms are suggested by family history.
Pathophysiology
Individuals with Ehlers-Danlos syndrome (EDS) demonstrate connective-tissue abnormalities due to defects in the inherent strength, elasticity, integrity, and healing properties of the tissues. [11] The specific characteristics of a particular form of EDS stem from the tissue-specific distribution of various components of the extracellular matrix (ECM). The ECM is defined as the outer cell components of tissue that provide structural support to the cells; it is the distinguishing feature of connective tissue. Each tissue and organ system has an array of connective proteins. Unique to each connective protein array is the path of production, its relative proportion, and distribution in tissues or organs. In addition, the defined interactions of various components of the matrix are tissue specific.
Major constituents of the extracellular matrix
Ehlers-Danlos syndrome (EDS) is caused by various abnormalities in the synthesis and metabolism of collagen and other connective-tissue proteins in the ECM, such as elastin, proteoglycans, and macromolecular proteins.
Collagen is the most abundant protein in the body and is the most common protein found in the ECM. [12] Collagen proteins are multimeric, occurring in trimers with a central triple helical region. A minimum of 29 genes contribute to collagen protein structure, and these genes are located on 15 different human chromosomes, genetically coding for no less than 19 identifiable forms of collagen molecules.
Elastins, in contrast to the structural support of collagens, give elasticity to the tissues. Elastin allows for the tissues to stretch and return to their original state and hence is present in the ECM of blood vessels, lungs, and skin. [13] Elastic fibers are created by the association of elastin with an underlying microfibrillar array. The underlying basis of all connective-tissue matrices is the microfibrillar array. An example of a microfibrillar protein is fibrillin. Fibrillin-1 gene mutations on chromosome 15 produce an abnormal fibrillin, as is characteristic of patients with Marfan syndrome.
Elastin and other structural proteins are woven onto the microfibrillar array to provide the basic meshwork for the connective-tissue matrix. Abnormalities of elastin have been associated with other connective-tissue disorders such as cutis laxa. Deletion of the elastin gene occurs in patients with Williams syndrome.
Proteoglycans are core proteins that are bound to glycosaminoglycans (also commonly termed mucopolysaccharides). Essentially, proteoglycans are the glue of the connective-tissue protein that seal and cement the underlying connective-tissue matrix.
Macromolecular proteins include the glycoproteins of the basement membrane (type IV collagen, laminin, entactin) and the ECM (fibronectin, tenascin).
Epidemiology
Frequency
EDS seems to have an overall prevalence of at least 1 in 5000 persons. The hypermobility and classical forms of EDS are the most common, with the former having a prevalence of perhaps 1 in 5,000 to 1 in 20,000 people, and the classical type being found in perhaps 1 in 20,000 to 1 in 40,000 individuals. [14]
Mortality/Morbidity
Reduced life expectancy is not generally a feature of Ehlers-Danlos syndrome (EDS), with the exception of vascular-type EDS (type IV). Median life expectancy for patients with vascular-type EDS is 50 years because medium-sized arteries, and the gastrointestinal tract, can spontaneously rupture. Uterine rupture during pregnancy has also been reported.
Morbidity in EDS is related to the primary pathophysiology and includes dislocations, pain, or both from chronic joint laxity and instability. Aberrant wound healing and scarring due to abnormal tensile strength of the skin also happens. [15] Rectal prolapse can occur, as described in the classical-type EDS (types I and II) patients. [16]
A study by Kim et al using the 2000-2012 National Inpatient Sample (NIS) indicated that compared with controls, hospitalized patients with EDS have a significantly greater likelihood of suffering from various cerebrovascular conditions, including carotid dissection, vertebral dissection, cervical artery aneurysm, cerebral aneurysm, and cerebrovascular malformation. [17]
In a retrospective study of 144 patients with vascular-type EDS, Adham et al found that 82 of them (56.9%) had a supra-aortic trunk (SAT) lesion, with 64.6% of the lesion patients being female. The SAT lesions were seen primarily in the internal carotid and vertebral arteries (56.3% and 28.9% of the lesions, respectively). Sixteen of the 82 patients (19.5%) were reported to have suffered transient ischemic attack or stroke. [18]
Race
Ehlers-Danlos syndrome equally affects all races.
Sex
Of the 6 major types of Ehlers-Danlos syndrome classified by Villefranche nosology, both males and females are equally affected, as the genetic coding causing the differing phenotypes are located on the autosomes (chromosomes 1-22) and not the sex chromosomes (X or Y).
Age
Ehlers-Danlos syndrome is a genetic disorder. As such, this syndrome and its various types are present at birth; however, symptoms may not become apparent until later in life.
-
Patient with Ehlers-Danlos syndrome. Note the abnormal ability to elevate the right toe. Courtesy of Enrico Ceccolini, MD.
-
Girl with Ehlers-Danlos syndrome. Dorsiflexion of all the fingers is easy and absolutely painless. Courtesy of Enrico Ceccolini, MD.
-
Patient with Ehlers-Danlos syndrome mitis. Joint hypermobility is less intense than with other conditions. Courtesy of Enrico Ceccolini, MD.
Tables
Type |
Inheritance |
Previous Nomenclature |
Major Diagnostic Criteria |
Minor Diagnostic Criteria |
Classical |
Autosomal dominant |
Types I and II |
Marked skin hyperextensibility, wide atrophic scars, joint hypermobility |
Smooth, velvety skin; easy bruising; tissue fragility; molluscoid pseudotumors (calcified hematomas over pressure points, eg, elbows); subcutaneous spheroids (fat-containing cysts on forearms and shins); joint hypermobility (eg, sprains, dislocations, subluxations); flat feet; muscle hypotonia; gross motor delays; postoperative complications (eg, hernia); manifestations of tissue fragility (eg, hiatal hernia, anal prolapse, cervical insufficiency); positive family history |
Hypermobility |
Autosomal dominant |
Type III |
Generalized joint hypermobility, affecting both large (elbows, knees) and small (fingers, toes) joints; skin involvement (soft, smooth and velvety) |
Recurrent joint dislocations and subluxations of shoulder, patella, and temporomandibular joints; chronic joint pain; limb pain; musculoskeletal pain; bruising tendencies; positive family history |
Vascular *Considered most serious EDS type, owing to risk of spontaneous arterial or organ rupture |
Autosomal dominant |
Type IV |
Thin, translucent skin, easy to see vasculature through the skin, especially chest and abdomen; arterial/intestinal fragility or rupture; extensive bruising with minor trauma; characteristic facial appearance of large eyes, thin nose, lobeless ears; short stature; thin scalp hair |
Acrogeria, aging skin; decrease of subcutaneous tissue in the face and extremities; gingival recession; hypermobile small joints; tendon/muscle rupture; clubfoot; early onset varicose veins; arteriovenous fistula; carotid-cavernous fistula; pulmonary conditions, pneumothorax, pneumohemothorax; positive family history; sudden death in close relative |
Kyphoscoliosis |
Autosomal recessive |
Type VI (Lysyl hydroxylase deficiency-collagen-modifying enzyme) |
Generalized joint laxity; severe hypotonia at birth; delayed gross motor development; progressive scoliosis (present at birth); scleral fragility or ocular globe rupture post minor trauma |
Tissue fragility; atrophic scars; easy bruising; spontaneous arterial rupture; marfanoid habitus; microcornea; osteopenia; positive family history (affected sibling) |
Arthrochalasia |
Autosomal dominant |
Types VIIA and VIIB |
Congenital hip dislocation; severe generalized joint hypermobility; recurrent subluxations |
Skin hyperextensibility with easy bruising; tissue fragility with atrophic scars; muscle hypotonia; kyphoscoliosis, mild osteopenia |
Dermatosparaxis |
Autosomal recessive |
Type VIIC |
Severe skin fragility; marked bruising; saggy, redundant skin, especially of the face; scars not atrophic |
Soft, doughy skin; premature rupture of membranes; hernias (umbilical and inguinal) |
Type |
Old Nomenclature |
Protein Abnormality |
Gene Abnormality |
Chromosome Locus |
Classical |
Types I and II |
Type V collagen |
*COL5A1,COL5A2 COL1A1 |
*9q34.3 2q32.3 17q21.3 |
Hypermobility |
Type III |
Type III collagen Tenascin-XB |
COL3A1 TNXB |
2q32.2 6p21.3 |
Vascular |
Type IV |
Type III collagen |
COL3A1 |
2q32.2 |
Kyphoscoliosis |
Type VI |
Lysyl hydroxylase deficiency (some) |
PLOD1 |
1p36.22 |
Arthrochalasia |
Types VIIA and VIIB |
Type I collagen |
A: COL1A1 B: COL1A2 |
17q21.33 7q21.3 |
Dermatosparaxis |
Type VIIC |
N-proteinase |
ADAMTS2 |
5q35.3 |









