Practice Essentials
In 1963, Lejeune et al described a syndrome consisting of multiple congenital anomalies, intellectual disability, microcephaly, abnormal face, and a mewing cry in infants with a deletion of a B group chromosome (Bp-), later identified as 5p-. [1]
Cri-du-chat syndrome is an autosomal deletion syndrome caused by a partial deletion of the p arm of chromosome 5 (5p) and is characterized by a distinctive, high-pitched, catlike cry in infancy, with growth failure, microcephaly, facial abnormalities, and intellectual disablity throughout life. [2] See the images below.
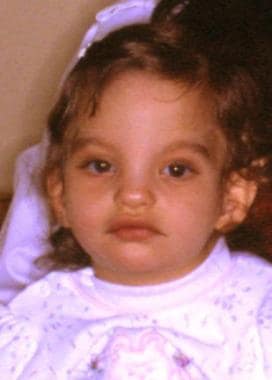 Infant with cri-du-chat syndrome. Note the round face with full cheeks, hypertelorism, epicanthal folds, and apparently low-set ears.
Infant with cri-du-chat syndrome. Note the round face with full cheeks, hypertelorism, epicanthal folds, and apparently low-set ears.
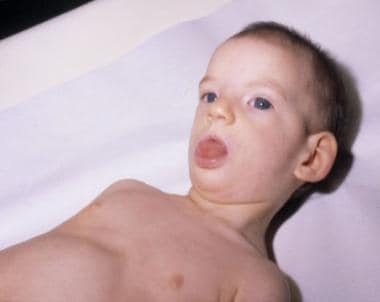 Child with cri-du-chat syndrome. Note the hypertonicity, small and narrow face, dropped jaw, and open-mouth expression secondary to facial laxity.
Child with cri-du-chat syndrome. Note the hypertonicity, small and narrow face, dropped jaw, and open-mouth expression secondary to facial laxity.
In recent years, the application of genetic molecular methods introduced advances in the diagnosis and typification of the cri-du-chat syndrome. [3]
Signs and symptoms of cri-du-chat syndrome
Newborns have a characteristic mewing cry, a high-pitched, monochromatic cry that is considered pathognomonic for this syndrome.
Neonatal complications include poor sucking, need for incubator care, respiratory distress, jaundice, pneumonia, and dehydration.
In addition, common neonatal findings include the following:
-
Low birth weight
-
Hypotonia
-
Microcephaly
-
Growth retardation
-
Round face with full cheeks
-
Hypertelorism
-
Epicanthal folds
-
Down-slanting palpebral fissures
-
Strabismus
-
Flat nasal bridge
-
Down-turned mouth
-
Micrognathia
-
Low-set ears
-
Short fingers
-
Single palmar creases
-
Cardiac defects
Findings in childhood include the following:
-
Severe intellectual disability
-
Developmental delay
-
Microcephaly
-
Hpertonicity
-
Premature graying of the hair
-
Small, narrow, and often asymmetrical face
-
Dropped-jaw, open-mouth expression secondary to facial laxity
-
Short philtrum
-
Malocclusion of the teeth
-
Scoliosis
-
Short third-fifth metacarpals
Children with cri-du-chat syndrome also have chronic medical problems such as upper respiratory tract infections, otitis media, severe constipation, and hyperactivity.
Workup in cri-du-chat syndrome
Laboratory tests include the following:
-
Conventional cytogenetic studies
-
High-resolution cytogenetic studies
-
Fluorescence in situ hybridization (FISH)
-
Chromosome comparative genomic hybridization (CGH)
-
Microarray CGH
-
Single-nucleotide polymorphism (SNP)–based prenatal test
Imaging studies include skeletal radiography, magnetic resonance imaging (MRI), and echocardiography.
Management of cri-du-chat syndrome
Care is supportive. No specific treatment is available for cri-du-chat syndrome. [4] Chronic medical problems such as upper respiratory tract infections, otitis media, and severe constipation require appropriate treatment.
Correction of congenital heart defects may be indicated. Medical problems involving minor malformations such as strabismus and clubfoot may be amenable to surgical correction. Orchiopexy may be necessary in patients with undescended testes.
Pathophysiology
The characteristic cry is perceptually and acoustically similar to the mewing of kittens. This unusual cry is due to structural abnormalities of the larynx (eg, laryngeal hypoplasia) and CNS dysfunction. The laryngeal appearance may be normal or may exhibit marked anatomical abnormalities such as floppy epiglottis, small larynx, and asymmetrical vocal cords. However, the cause of the characteristic cry cannot be entirely ascribed to the larynx. A developmental field may connect the brain and the affected clivus region of the cranial base with the laryngeal region from which the characteristic cry derives. This area of the brain is probably deformed in patients with cri-du-chat syndrome. The characteristic cry usually disappears over time.
Cri-du-chat syndrome is caused by a partial or total deletion of genetic material on the short arm of chromosome 5. The size of the deletion could affect from region 5p15.3 to the complete loss of the short arm. Most of the cases (approximately 80%) are due to a de novo deletion, a little more than 10% of the cases are originated by a parental translocation, and less than 10% are associated with cytogenetic rare aberrations. [5, 6]
Genotype-phenotype studies in cri-du-chat syndrome led to the identification of two separate chromosomal regions, hemizygosity for which is associated with specific phenotypes. [7]
A deletion of 5p15.3 results in the manifestation of a catlike cry, [8] whereas a deletion of 5p15.2 results in the presentation of the other major clinical features of the syndrome. [9] Moreover, a region for speech delay in 5p15.3 has been identified. [10]
Epidemiology
Frequency
United States
The estimated prevalence is about 1 in 50,000 live births. The prevalence among individuals with intellectual disability is about 1.5 in 1000.
Mortality/Morbidity
With contemporary interventions, the chance of survival to adulthood is possible. Currently, the mortality rate of cri-du-chat syndrome is 6-8% in the overall population.
Pneumonia, aspiration pneumonia, congenital heart defects, and respiratory distress syndrome are the most common causes of death.
A study by Sanders et al indicated that symptoms of primary ciliary dyskinesia (PCD), another disorder traced to chromosome arm 5p, are prevalent in patients with cri-du-chat syndrome. Symptoms associated with PCD found in the cri-du-chat syndrome patients included unexplained neonatal distress (35%), year-round nasal congestion that arose in infancy (32%), and a year-round, wet cough that started in infancy (20%). [11]
A study by Lefranc et al indicated that protein-energy malnutrition is a frequent problem in cri-du-chat syndrome, with a review of questionnaires from 36 families revealing evidence of such malnutrition in 47% of patients. [12]
Race
No racial predilection has been found.
Sex
A significant female predominance is observed in affected newborns, with a male-to-female ratio of 0.72:1.
Age
The condition is detected in newborns and infants because of the catlike cry and dysmorphic features.
-
Infant with cri-du-chat syndrome. Note the round face with full cheeks, hypertelorism, epicanthal folds, and apparently low-set ears.
-
Child with cri-du-chat syndrome. Note the hypertonicity, small and narrow face, dropped jaw, and open-mouth expression secondary to facial laxity.
-
Fluorescent in situ hybridization (FISH) study of a patient with cri-du-chat syndrome. FISH photograph shows deletion of a locus-specific probe for the cri-du-chat region. Spectrum orange color represents chromosome 5–specific signal and spectrum green is cri-du-chat locus signal. Absence of a green signal indicates monosomy for that region (left, interphase cell; right, metaphase chromosome spread).
-
G-banded karyotype [46,XX,del(5)(p13)].
-
G-banded karyotype of a carrier father [46,XY,t(5;17)(p13.3;p13)].





