Practice Essentials
Cockayne syndrome (CS) refers to the spectrum that includes:
-
Cockayne syndrome type I, the classic form (also referred to as the moderate form)
-
Cockayne syndrome type II, a more severe form with symptoms present at birth; the clinical features of Cockayne syndrome type II overlap with those of cerebro-oculo-facio-skeletal (COFS) syndrome, which is also called Pena-Shokeir syndrome type II
-
Cockayne syndrome type III, a milder form
-
Xeroderma pigmentosa–Cockayne syndrome (XP-CS).
However, the discussion in this article is limited to Cockayne syndrome types I and II, also termed Cockayne syndrome types A and B, respectively.
Cockayne syndrome type I (CKN1; Online Mendelian Inheritance in Man [OMIM] number 216400) and Cockayne syndrome type II (CSB; OMIM number 133540) are rare autosomal recessive disorders that feature growth deficiency, neurologic dysfunction, premature aging, and pigmentary retinal degeneration, along with a complement of other clinical findings. Cockayne syndrome type II presents at birth, with death within the first decade, whereas Cockayne syndrome type I appears during early childhood, with death occurring in early adolescence, although there are reports of some patients surviving until early adulthood. Cockayne syndrome was first reported in 1936, by Edward Alfred Cockayne. [1, 2]
Signs and symptoms of Cockayne syndrome
Characteristics of Cockayne syndrome include the following:
-
Delayed psychomotor development
-
Growth failure
-
Poor feeding
-
Photosensitive rashes; decreased amounts of subcutaneous tissue; dry, scaly skin; and thin, dry hair
-
Pigmentary degeneration of the retina
-
Cataracts
-
Increased or decreased muscle tone and reflexes
-
Mild-to-severe sensorineural hearing loss
-
Moderate-to-severe dental caries; permanent teeth have short roots
-
Cryptorchidism or testicular hypoplasia
Workup in Cockayne syndrome
The evaluation includes the following:
-
Genetic evaluation
-
Developmental evaluation
-
Ophthalmologic evaluation
-
Neurologic evaluation
-
Gastrointestinal evaluation with a nutritionist
-
Audiologic evaluation
-
Dermatologic evaluation
-
Dental evaluation
Computed tomography– (CT-) or magnetic resonance imaging– (MRI-) scan findings include increased ventricular size, cerebral atrophy, white matter abnormalities, and normal pressure hydrocephaly.
Skeletal radiographs depict vertebral body and pelvic abnormalities.
Management of Cockayne syndrome
Various management strategies include the following:
-
Physical therapy - Helps to prevent contractures and maintain ambulation
-
Feeding therapy - Including consideration of gastrostomy tube for failure to thrive
-
Management of hearing loss – Such as through hearing aids or other devices, if necessary
-
Evaluation for and, if necessary, treatment of cataracts
-
Administration of antiseizure and antispasticity medications, if necessary
-
Limitation of ultraviolet (UV) radiation exposure - Sunscreen should be applied liberally, and excessive sun exposure should be avoided; sunglasses will help with the photosensitivity of the eyes
Pathophysiology
Cockayne syndrome type I is characterized by normal growth parameters at birth. However, within the first 2 years of life, growth and development become abnormal and continue to progress as such. Height, weight, and head circumference fall below the fifth percentile, while impairment of vision, hearing, and neurologic function leads to severe disability. The characteristic physical appearance of cachectic dwarfism, with thinning of the skin and hair, sunken eyes, and a stooped standing posture, illustrates the aging process. Pathologic studies reveal diffuse and extensive demyelination in the central and peripheral nervous systems. Patients demonstrate pericapillary calcifications in the cortex and basal ganglia at an early age; severe neuronal loss in the cerebral cortex and cerebellum also occurs. These changes correlate with the physiologic changes of aging and the significant degree of neurologic dysfunction. Pigmentary retinopathy, photosensitivity, and other features, such as dental anomalies, vision loss, and hearing loss, also occur.
Cockayne syndrome type II is referred to as severe Cockayne syndrome. Children with this condition are found at birth to have growth failure. Neurologic development is static, with very little progress. Contractures of the spine and joints, known as arthrogryposis, are present. Other features overlap with those of Cockayne syndrome type I. Death occurs on average by age 7 years.
Epidemiology
Frequency
United States
The incidence of Cockayne syndrome is estimated to be 2.7 cases per million births in the United States and Western Europe. [3]
International
Similarly, a study by Kubota et al found the incidence of Cockayne syndrome in Japan to be an estimated 2.77 cases per million births. [4] There have also been reported areas in Israel with high carrier rates. [3]
Mortality/Morbidity
Patients are at risk for postnatal growth failure, neurologic deterioration, pigmentary retinal degeneration, and premature death before adulthood. Details are as follows:
-
Postnatal growth failure - Profound growth failure begins within the first year of life. Weight is affected more than length, and cachectic dwarfism results, with microcephaly; a rare subset of patients, classified as having severe Cockayne syndrome type II, have low birth weight with almost no postnatal growth
-
Neurologic deterioration - Deterioration of the central and peripheral nervous system leads to spasticity, ataxia, tremor, seizures, intellectual disabilities, muscle atrophy, and weak cry and feeding
-
Pigmentary retinal degeneration - This diagnostic criterion for Cockayne syndrome type I (salt-and-pepper appearance in the retinas) develops later in childhood; cataracts are the second most common eye finding
-
Premature death - The characteristic appearance of aging in children with Cockayne syndrome is striking; the mean and median age of death is 12 years, and most patients die as a result of pneumonia or other respiratory infections
Race
Cockayne syndrome type I is panethnic.
Sex
The male-to-female ratio is 1:1, which is consistent with an autosomal recessive disorder. The pattern of autosomal recessive inheritance is illustrated in the image below.
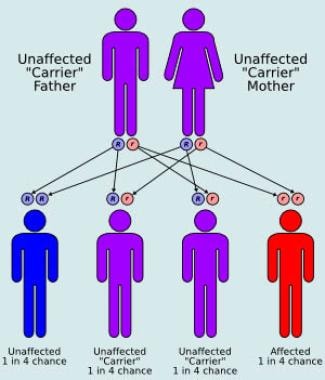 Autosomal recessive inheritance pattern.
Autosomal recessive inheritance pattern.
Age
This is a progressive congenital genetic disorder. Cockayne syndrome type I usually becomes evident within the first 2 years of life, when growth and development are notably delayed. Cockayne syndrome type II is usually evident at birth, with growth delays, lack of developmental progress, and congenital cataracts. [5]
Prognosis
In persons with Cockayne syndrome type I, the mean age of death is 12 years, with very few individuals surviving into the third decade. In the type II form, the mean age of death is 7 years. [3]
-
Autosomal recessive inheritance pattern.








