Background
Chromosomal breakage syndromes are a group of genetic disorders that are typically transmitted in an autosomal recessive mode of inheritance. In culture, cells from affected individuals exhibit elevated rates of chromosomal breakage or instability, leading to chromosomal rearrangements. The disorders are characterized by a defect in DNA repair mechanisms or genomic stability, and patients with these disorders show increased predisposition to cancer.
The following specific chromosome breakage syndromes are addressed in separate subsections of this article:
Chromosomal breakage syndromes are relatively rare; most practicing physicians may never see a patient with a chromosomal breakage syndrome. Some of the specific syndromes occur at relatively high rates in certain ethnic groups. Diagnosis is complicated because the symptoms may be varied and complex. These disorders are often lethal.
Patients with ataxia telangiectasia, also known as Louis-Bar syndrome, are hypersensitive to ionizing radiation, while patients with Bloom syndrome, Fanconi anemia, and xeroderma pigmentosum are sensitive to UV radiation. The ataxia telangiectasia Rad3–related (ATR) protein responds to UV damage, whereas the ataxia telangiectasia mutated (ATM) protein responds to double-strand breaks (DSBs) caused by ionizing radiation and radiomimetic compounds.
Table 1 provides a summary outline of the gene symbols, chromosomal locations, radiation sensitivity characteristics, immunodeficiencies, chromosome breakage characteristics, and major cancer risk for each of these disorders.
Table 1. Chromosomal Breakage Syndromes With Neoplasias Caused by Defective DNA Repair (Open Table in a new window)
Syndrome |
Gene Symbol: Chromosome Band |
Hypersensitivity: Immunodeficiencies |
Chromosome Breakage |
Cancer Risk |
Ataxia telangiectasia |
ATM: 11q22.3 (11q22–11q23 Human Genome Organization) |
Roentgentherapy, ionizing radiation, radiomimetic compounds: decreased immunoglobulin (Ig)A, IgG2, IgG, IgE |
TCRA: 14q11 TCRB: 7q35 IGH: 14q32 No increased sister chromatid exchanges (SCE), but increased gaps and breaks, nonhomologous interchanges |
The risk of neoplasia is 38% (85% are leukemias and lymphomas [B-cell]). Young children have acute lymphoblastic leukemia of T-cell origin. Older children have T-cell leukemia. The risk for other cancers is increased 4-fold (2-fold to 3-fold increased risk for breast cancer in carriers). |
Bloom syndrome < 1% of mutations |
BLM: 15q26.1 |
UV, cancer chemotherapeutic agents: Decreased IgA and IgM, and/or IgG |
SCE 12-15 times higher, homologous interchanges, increased gaps and breaks |
The risk for leukemia, lymphoma, adenocarcinoma, and other cancers is increased 5-fold to 8-fold. |
Fanconi anemia |
FANCA: 16q24.3 FANCB: Xp22.31 FANCC: 9q22.3 FANCD1/BRCA2: 13q12.3 FANCD2: 3p25.3 FANCE: 6p22-p21 FANCF: 11p15 FANCG/XRCC9: 9p13 FANCH = FANCA FANCI: 15q25-q26 FANCJ/BRIP1/BACH1: 17q23 FANCL/FAAP43: 2p16.1 FANCM/KIAA1596: 14q21.3 FANCN/PALB2: 16p12 FANCO/RAD51C: 17q22 FANCP/SLX4: 16p13.3 |
UV, chemotherapeutic agents for immunosuppression, diepoxybutane, mitomycin C, other DNA-crosslinking agents |
Increased gaps and breaks, 30% between nonhomologous chromosomes, clones with translocations |
For aplastic anemia, the risk is increased for pancytopenia, leukemia, acute myeloid leukemia, squamous cell carcinoma, medulloblastoma, Wilms tumor, breast cancer, and liver cancer. |
Xeroderma pigmentosum |
XPA: 9q22.3 XPB/ERCC3: 2q21 XPC: 3p25 XPD/ERCC2: 19q13.2-q13.3 XPE/DDB2: 11p12-p11 XPF/ERCC4: 16p13.3-p13.13 XPG/ERCC5: 13q33 XPV/POLH: 6p21.1-p12 ERCC1: 19q13.2-q13.3 |
UV XPG group may be sensitive to X-rays |
Increased gaps and breaks, increased SCE after UV |
The risk is increased 1000-fold for squamous cell carcinoma, basal cell carcinoma, malignant melanoma, and fibrosarcoma and is increased 10-fold to 20-fold for other tumors. |
Pathophysiology
Most of the genes in these disorders were identified because of searches for the disorders based on either the consequences or causes of the symptoms.
These syndromes are characterized by different types of cancer, including leukemia, lymphoma, and solid tumors (eg, breast cancer, skin cancer). In addition, the syndromes are associated with other traits, such as cerebellar ataxia, immunodeficiencies, growth retardation, microcephaly (not in ataxia telangiectasia), skeletal abnormalities, hypogonadism, pancytopenia, and abnormal pigmentation. Complications in these disorders make therapy difficult. State-of-the-art testing and supportive medical care can extend a patient's life span. An accurate genetic diagnosis is essential, as is providing effective genetic counseling to families.
Ataxia Telangiectasia
Ataxia telangiectasia, also known as Louis-Bar syndrome, is a complex hereditary syndrome first described in 1941 by French physician Denise Louis-Bar. The chromosomal instability was identified in 1966, clinical features were defined in the 1970s, and the gene responsible for ataxia telangiectasia was discovered in 1995.
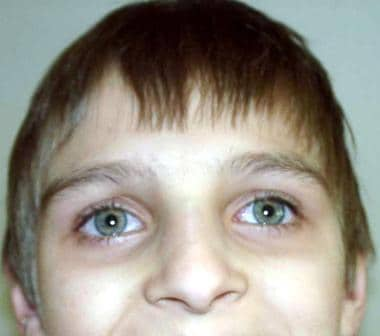
Ataxia telangiectasia is an early onset autosomal recessive disorder characterized by cerebellar ataxia, telangiectasias, immunodeficiency, hypogonadism, and predisposition to neoplasias. Patients are particularly sensitive to ionizing radiation and radiomimetic compounds.
The relevant gene is known by the symbol ATM, for ataxia telangiectasia mutated. The normal product of the gene is a serine/threonine kinase, which is activated by double-strand breaks in DNA in G2 of the cell cycle. Once activated, the kinase phosphorylates over 40 substrates in the signaling cascade involved in repair of double-strand breaks in DNA. Cells with deficient ATM function fail to repair double-strand breaks in DNA, which leads to chromosomal instability, chromosome breaks, and characteristic chromosome rearrangements.
Pathophysiology
Progressive cerebellar ataxia is usually the first symptom of ataxia telangiectasia observed, becoming apparent in children aged 1–4 years (average age of onset 2 y). It is probably the most common progressive cerebellar ataxia of early childhood. The cerebellum atrophies early, with loss of Purkinje cells and granule cells. An affected child may exhibit staggering when first learning how to walk. However, this truncal ataxia improves from age 2-4 years, followed by slurred speech and oculomotor apraxia (horizontal and vertical eye movements). Choreoathetosis is found in almost all affected patients, and myoclonic jerking and intention tremors are noted in 25%.
The most striking feature of the disease is the cephalooculocutaneous telangiectasia, appearing first in bulbar conjunctivae, next in the ears, cheeks, neck, antecubital fossae, hands, and knees. Telangiectasias appear in children aged 2-8 years (average age of onset 3.6 y). The ataxia and telangiectasias tend to be progressive until children reach their mid teen years. Other symptoms are immunodeficiency, premature aging including canities, degeneration of thymus and gonads, disuse atrophy of the muscles, and growth retardation. Some patients present with abnormal liver function, hyperglycemia, insulin-resistant diabetes, or a combination of these.
Patients become wheelchair-bound in their teens, and require assistance in performing the activities of daily living.
People with ataxia telangiectasia usually have normal intelligence; many American and British patients graduated from high school and some finished college or graduate studies. Intellectual disability is uncommon.
Patients are susceptible to tumors and neoplasias (seen in 38%); leukemia and lymphoma account for about 85% of these malignancies. Younger children often have acute lymphoblastic leukemia of T-cell origin, and older children often have aggressive T-cell leukemia. Lymphomas usually involve B cells. When patients are older, sarcomas, solid tumors, and carcinomas occur at 4 times the incidence in the general population. Female heterozygotes (carriers) of an ATM mutation have a 2-fold to 3-fold increased risk of breast cancer, which occurs predominantly in premenopausal women. Loss of heterozygosity of ATM (loss of the normal allele, presence of only the mutated allele) was reported in 30–40% of patients with breast tumors, and 50–70% had altered ATM protein levels.
Because patients with ataxia telangiectasia are hypersensitive to ionizing radiation and radiomimetic compounds, chemotherapy or radiation therapy normally used to treat cancers in other patients may have deleterious and even lethal effects.
Patients have unusually high rates of recurrent sinopulmonary viral or bacterial infections. Infection incidences may increase during the first year of life, but infections do not become common until children are aged 3-8 years. Defective cellular and humoral immunity occurs in 60-80% of patients. levels of IgA, IgG2, and IgE tend to be low or absent.
Serum alpha-fetoprotein is elevated; however, unaffected infants may have abnormally high levels until they are aged 2 years.
Patients may have stunted growth. Their body mass index (BMI) is low despite adequate nutrition; one study reported that serum levels of insulin-like growth factor-1 (IGF-I) were below the third percentile in 56% of patients, and levels of insulin-like growth factor–binding protein-3 (IGFBP-3) were below the third percentile in 81% of patients. Many patients have testicular and ovarian hypoplasia, and, in some cases, the ovaries may be totally absent.
A study by Natale et al found that patients with classic ataxia telangiectasia continuously falter with regard to height, weight, and body mass index (BMI) after reaching age 2 years. Eventually, according to the investigators, their growth parameters decline to a level at or below the third centile, as determined by the Centers for Disease Control and Prevention (CDC). The reduction for median height was found to be most pronounced in girls, but after age 15 years, females in the study experienced a recovery in height centiles. [3]
A retrospective study by Nissenkorn et al suggested that growth abnormality in ataxia telangiectasia is a primary condition, not a phenomenon secondary to nutritional impairment or disease severity. In the study, of 52 patients with ataxia telangiectasia, the investigators found that patients’ median height standard deviation score (SDS) was below normal in infancy and remained so up to follow-up in adulthood, with the patients’ weight SDS being less impaired than height SDS up to age 4 years. IGF-I SDS, although low, had no correlation with height SDS, according to the study. [4]
A milder form of ataxia telangiectasia, with later onset of neurologic progression and decreased radiosensitivity, was reported in patients who retained some ATM protein kinase activity. [5] Affected siblings in the same family may demonstrate variable severity and different clinical and immunological abnormalities. [6, 7] Other patients presented with mild symptoms despite lack of protein for ATM, severely defective DNA-damage control response, and marked cerebral atrophy. These findings suggest the presence of modifier genes and environmental factors, including Epstein-Barr virus and other pathogens, that influence the onset and severity of neurodegeneration and other symptoms.
The mothers of affected patients have an increased risk of breast cancer. [8] Ataxia telangiectasia carriers may also be at an increased risk for heart disease.
Patients with ataxia telangiectasia generally do not reproduce.
Frequency
Ataxia telangiectasia is a very rare disorder; it occurs at a frequency of 1 case per 40,000-100,000 live births worldwide. The frequency of carriers is -1.8%, an especially significant rate because of their increased sensitivity to ionizing radiation and breast cancer.
Mortality/morbidity
Patients become wheelchair-bound by age 10-15 years. Severely affected patients usually do not survive childhood. Pneumonia and lymphoreticular cancer in adolescence were once common causes of death. Over the last 2 decades, the expected lifespan has increased significantly; most patients now live beyond 25 years, and some live into their fifth and sixth decades. Pulmonary disease is still the leading cause of death.
Sex
Males and females are affected equally.
Age
Ataxia initially presents in children aged 1-4 years, usually at the onset of walking (which tends to be somewhat delayed in affected individuals). Telangiectasias may be concurrent or appear later, in children aged 2-8 years. Malignancies occur early.
Genetic causes
Ataxia telangiectasia is caused by a single gene inherited as an autosomal recessive trait. Patients are usually compound heterozygotes, with different mutations in the 2 alleles of ATM. However, some ATM mutations, especially missense mutations, produce a dominant negative effect (in which a mutation in only 1 of the 2 alleles of the gene results in the disease phenotype). Chromosome gaps, breaks, and interchanges between nonhomologous chromosomes are a result of the defective DNA damage repair.
The gene symbol is ATM (includes complementation groups A, C, and D). The ATM locus is at 11q22.3 (11q22–11q23 according to the Human Genome Organization, HUGO). It comprises more than 150 kilobases (kb) and 66 exons (62 coding). More than 500 unique mutations have been reported; [9] most result in lack of protein for ATM. No hotspots of mutations were found. Some common mutations are listed in Table 2.
The protein has 370 kDal and consists of 3081 amino acids. It is a serine/threonine kinase related to phosphoinositide-3 kinase (PI3K) and is found in the cell nucleus. ATM is involved in oxidative stress, cell cycle control, and DNA-damage repair. It is a tumor suppressor gene that responds to double-strand breaks in DNA and directly or indirectly phosphorylates over 40 substrates to promote end-joining DNA repair.
Table 2. Ataxia Telangiectasia Populations, ATM Mutations, and Effects (Open Table in a new window)
Populations Affected |
ATM Mutation |
Effect or Comments |
|
Worldwide Amish, Turkish, Italian, German, Brazilian, Polish |
1563delAG |
Most common |
|
African American, and Korean |
L546V |
Increased risk of breast cancer |
|
American |
7271T>G5762ins137nt 8494C>T 5762ins137nt |
Kinase-dead mutation, milder phenotype Milder phenotype Slower neurologic deterioration |
|
Chinese |
G449A G204X R2227C |
Homozygous |
|
English/Irish, Polish |
7010delGT |
... |
|
German, Polish, American-Hispanic |
IVS20–579delAAGT |
... |
|
Iranian and Polish |
381delA |
Truncated protein in 23 of 25 carriers |
|
Italian, Polish |
8545C>T |
... |
|
Japanese, Polish |
742C>T |
... |
|
Korean |
IVS21+1049T>C IVS34+60G>A 3393T>G |
Increased risk of breast cancer, especially in premenopausal women; a genotypic polymorphism may play a role in breast cancer |
|
Mennonite, Polish, Danish, Norwegian, American Hispanic, German, Russian |
5932G>T |
... |
|
Philippine, Turkish, Polish |
5712insA |
... |
|
Polish, Danish, American Hispanic, Brazilian, Portuguese |
IVS53–2A>C |
Most common Polish mutation |
|
Polish, Swedish, German, French |
6095G>A |
Most common Polish mutation |
|
Spanish, Polish |
5188C>T |
... |
|
Childhood T-cell acute lymphoblastic leukemia |
S707P; F858L; P1054R; L1472W; Y1475C |
Found more often in T-cell acute lymphoblastic leukemia, with higher WBC count and increased risk of relapse |
|
Laboratory studies
The following laboratory studies are useful:
-
Immunoblotting for the protein for ATM is at present the preferred test for a diagnosis of ataxia telangiectasia. Of ataxia telangiectasia patients, 90% have no detectable protein for ATM, about 10% have trace amounts, and about 1% lack ATM protein kinase ("kinase dead"). Since the procedure requires at least 5 million cells or 25 mcg of lysate protein, a lymphoblastoid cell line (LCL) is established; this requires 4-6 weeks. If the ATM protein level is normal and the radiosensitivity is abnormal, the ATM kinase activity is tested. If the ATM protein level is higher than trace amounts and the radiosensitivity is normal, a diagnosis of ataxia telangiectasia is excluded.
-
ATM kinase activity can be assessed by immunoblotting of cell lysates and by using commercial antibodies to any phosphorylated ATM substrate. However, the kinase activity is difficult to quantify; it is much reduced in patients with a kinase-dead ATM mutation (7271T>G).
-
Radiosensitivity assay determines the survival of lymphoblastoid cells after irradiation with 1 Gy. This colony survival assay takes about 3 months. The assay results are abnormal in 99% of patients who had at least one identifiable ATM mutation. Because the ataxia telangiectasia cells cannot repair DNA damage efficiently, they are hypersensitive to the lethal effects of the irradiation. The same test possibly may provide prenatal ataxia telangiectasia detection, using chorionic villi samples. The test may also be used to identify individuals who do not have ataxia telangiectasia but may have another DNA repair disorder.
-
Cytogenetic analysis for chromosome breakage in dividing cells exposed to irradiation may be used to identify heterozygotes and chromosome aberrations in patients. Karyotyping is performed on peripheral blood. Lymphopenia and a decreased response to phytohemagglutinin hinder chromosome analysis, but 20-40% of chromosome breakage is observed in vitro. Persons with ataxia telangiectasia frequently have abnormalities involving chromosome 14, particularly a 7;14 chromosome translocation, which is seen in 5-15% of peripheral lymphocytes from patients that are stimulated with phytohemagglutinin and harvested at 72 hours. The chromosome breakpoints are typically 14q11 (T-cell receptor-alpha locus) and 14q32 (B-cell receptor locus). Prenatal diagnosis by chromosomal breakage studies were found to be unreliable in 3 cases.
-
Molecular genetic testing (DNA analysis) is performed to identify the ATM mutation, if the mutation is detected, the diagnosis of ataxia telangiectasia is confirmed. DNA analysis may also reveal a heterozygote (carrier). The test may be used for prenatal diagnosis; however, this can only be done if the mutations of both parents are known. Sequencing of the ATM coding region detects about 90% of its mutations. Linkage analysis may also identify carriers. Haplotype analysis can be done in certain ethnic groups and involves analyzing several short nucleotide sequences (about 5) at the ATM locus to identify founder mutations. The protein truncation test detects about 70% of ATM mutations.
-
Serum alpha-fetoprotein levels are elevated above 10 ng/mL in more than 95% of patients with ataxia telangiectasia. However, the levels may also remain above normal in unaffected children until age 2 years.
-
Serum immunoglobulin levels of IgA, IgG2 or total IgG, and IgE are decreased markedly or even absent.
-
Brain MRI can detect cerebellar atrophy; the cerebellum is visibly smaller by age 7 or 8 years.
-
Brain single-photon emission computed tomography (SPECT) may indicate cerebellar regional cerebral blood flow hypoperfusion.
The Web site for GeneTests lists laboratories that perform genetic testing for a wide variety of genetic disorders.
The work-up must include taking a family history.
Medical care
Antioxidants are recommended; however, no formal testing of their efficacy in patients with ataxia telangiectasia has been done. Studies are underway to measure the effects of antioxidant therapy on oxidative stress and metabolic abnormalities in affected patients. [10] In addition, a recent study recommends the placement of a gastrostomy tube at a young age, before the development of significant dysphagic, nutritional, and respiratory problems. [11] Early treatment with systemic corticosteroids was associated with clinical and radiographic improvement of chronic progressive interstitial lung disease.
Immunization to common pathogens, including pneumococcal conjugate and influenza vaccines, is recommended in all patients. Live viral vaccines, including MMR (measles, mumps, rubella), MMRV (measles, mumps, rubella, varicella), OPV (oral polio vaccine), and varicella, may be considered in all but the most severely lymphopenic patients. [12] Prophylactic azithromycin may also be considered. Patients with frequent and severe infections may benefit from intravenous immunoglobulin (IVIG) replacement therapy. Early and continuous physical therapy minimizes contractions. Supportive therapy may lessen drooling, choreoathetosis, and ataxia. Provision of a wheelchair may be necessary.
The European Workshop on Ataxia-Telangiectasia 2011 in Frankfurt, Germany produced a registry of approximately 60 patients and a discussion on the guidelines of patient care. [13]
Recommended referrals and consultations include the following:
-
Medical geneticists - For genetic diagnosis and genetic counseling
-
Neurologist - For specific problems associated with the progressive ataxia
-
Pulmonary/infectious disease specialist - For treatment of sinorespiratory infections (Progressive generalized bronchiectasis may not be responsive to conventional antibiotic management and may require aggressive pulmonary hygiene.)
-
Hematologist - For immunoglobulin therapy. (However, if no immunomodulatory therapy is undertaken and the patient does not present with severe recurrent infections, then the patient's immune status does not have to be routinely monitored.)
-
Oncologist - For monitoring and management of malignancies
For clinical trials search ClinicalTrials.gov.
Special concerns
Protect patients and carriers from exposure to ionizing and radiomimetic compounds.
The genetic nature of ataxia telangiectasia makes accurate genetic diagnosis and genetic counseling for family members essential. Counseling is most important for the carriers of an ATM mutation; although carriers may have a healthy appearance, patients and many carriers have an increased risk for cancer after exposure to radiation or radiomimetic compounds.
Because of advances in prenatal ataxia telangiectasia detection, refer carriers who are contemplating pregnancy to a genetics clinic. People with ataxia telangiectasia usually do not reproduce.
Affected individuals should be periodically monitored throughout life for early signs of a new malignancy. Parents of an affected child are obligate carriers and are at increased risk for cancer, especially breast cancer in females and coronary artery disease.
Cancer treatment should be modified to account for the higher risk of lethal side effects with conventional doses of radiotherapy and some radiomimetic chemotherapeutic agents.
Medical/legal pitfalls
The most common misdiagnosis is cerebral palsy. Friedreich ataxia and ataxia with oculomotor apraxia type 1 (AOA1) can be differentiated from ataxia telangiectasia by a radiosensitivity assay; these patients' lymphoblastoid cells are not radiosensitive. However, radiosensitivity is seen in patients with Nijmegen breakage syndrome, X-linked agammaglobulinemia, Fanconi anemia, ligase IV deficiency, Seckel syndrome, common variable immunodeficiency, and severe combined immunodeficiency. These disorders are not characterized by ataxia and elevated serum levels of alpha-fetoprotein.
Additional possible medicolegal pitfalls include (1) failure to counsel the parents of an affected child about recurrence risks in future pregnancies and (2) failure to advise the mother of an affected child about her increased risk of breast cancer and recommend high-risk breast cancer surveillance.
Bloom Syndrome
In 1954, David Bloom first described Bloom syndrome. In 1960, James German discovered disrupted and rearranged chromosomes in 0.5-11% of the cells of nearly 40 affected families. The autosomal recessive gene for Bloom syndrome, termed Bloom syndrome–mutated (BLM), has been mapped to chromosome band 15q26.1.
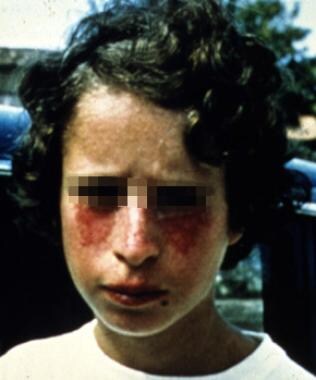 A young patient with Bloom syndrome showing the typical photodistributed erythema on the face. Courtesy of James L. German III, MD.
A young patient with Bloom syndrome showing the typical photodistributed erythema on the face. Courtesy of James L. German III, MD.
The main features of this syndrome are severe failure to thrive in infancy, stunted growth, small and narrow facies, sun-sensitive facial telangiectasias, immunodeficiency, and increased risk of malignancies. Growth retardation commences prenatally and continues postnatally. Infants experience feeding difficulties. The facial telangiectasias usually do not appear at birth but develop later after exposure to sunlight. Typically, the facial condition improves after childhood. Recurrent GI and respiratory tract problems occur in infancy. Patients tend to be infertile.
Pathophysiology
Growth retardation, the most conspicuous feature of Bloom syndrome, begins in utero and continues throughout the individual's lifetime; males reach an average height of 5 feet and females an average of 4 feet 9 inches. This growth retardation may be caused by the chromosomal abnormalities that lead to increased homozygosity of daughter cells.
Individuals have a dolichocephalic and microcephalic head shape, hypertrichosis, cheilitis, small narrow facies with telangiectasias, usually café-au-lait–colored macules in a butterfly pattern on the cheeks, although ears, forearms, and dorsal hands also may be involved. These telangiectasias vary in severity and are the result of hypersensitivity to sunlight, caused by a defective DNA helicase. Skin sensitivity to sunlight in patients with Bloom syndrome usually occurs within a few months of birth, an earlier age than in those affected by other similar syndromes (eg, ataxia telangiectasia, xeroderma pigmentosum).
Minor anatomic defects may be present (eg, malar hypoplasia, small mandible, absent upper lateral incisors, prominent ears and nose, syndactyly, polydactyly, and fifth finger clinodactyly). Patients tend to have a high-pitched voice and may present with bronchiectasis and chronic lung disease. Another feature seen is non–insulin-dependent diabetes mellitus.
Intelligence is usually unimpaired, but mild intellectual disability and learning disability were found in some patients.
Retinal hard drusen, or asymptomatic hyaline deposits located beneath the retinal pigment epithelium, was diagnosed in 2 patients with Bloom syndrome, one of these also had diabetic retinopathy and onset of leukemic retinopathy.
Azoospermia and cryptorchidism is found in males, and infertility and premature menopause are found in females.
Children with Bloom syndrome are predisposed to infection. Subsequently, a severe defect in immunity may result in life-threatening respiratory and GI infections. IgA and IgM levels are decreased, and IgG levels are occasionally decreased.
In a study involving 48 patients, 11 had malignancies. Of these, 2 had acute lymphocytic leukemia, 1 had squamous cell carcinoma of the esophagus, 1 had adenocarcinoma of the sigmoid colon, and 2 had cancer of the alimentary tract. Lymphomas were common, and cases of cervical carcinoma were reported. Patients have an impaired lymphocyte proliferation response to malignancy, and they are hypersensitive to chemotherapy.
Frequency
Bloom syndrome is extremely rare except in Ashkenazi Jews. German et al estimated a minimum gene frequency of 0.0042, which would correspond to an incidence of 1 per 55,000 Ashkenazim. [14] In 1999, Roa et al estimated the carrier frequency of the common mutation in approximately 97% of Ashkenazi Jewish patients at 1 in 194 persons. [15] This estimate corresponds to an incidence of 1 case per 40,000 persons of Ashkenazi Jewish descent.
In families of non-Jewish patients, consanguinity may be likely.
Mortality/morbidity
The Bloom syndrome registry maintained by German showed that 96% of the patients survived infancy and that most of the deaths (79%) were from malignancy, typically in the second or third decade of life. Patients with Bloom syndrome have a 1-in-8 risk of leukemia. One fourth of heterozygotes develop cancer at an early age.
Conventional cancer detection methods are used in Bloom patients; however the age to begin screening is much earlier that the general population. In persons younger than 20 years, leukemia is the main type of cancer. Colon cancer is the most common single cancer in patients with Bloom syndrome; screening should begin decades earlier than average and should be carried out more frequently than generally recommended.
Sex
Prevalence is slightly higher in males than in females; however, the reason for this difference is unknown.
Age
Shortness of stature is evident at birth, but the syndrome's typical facial erythema usually does not appear until several months after birth. [16] German reported first diagnoses of cancers in patients whose ages ranged from 4-46 years.
Causes
Frequency of sister chromatid exchanges (SCE) in homozygotes is high (ie, 12-15 times higher rate than reference range); the SCE rate in heterozygotes is normal.
Chromosome interchanges between homologous chromosomes, gaps, and breaks occur.
DNA repair after UV exposure is impaired.
Genetic causes can be summarized as follows:
-
Mode of inheritance is single gene, autosomal recessive.
-
The gene symbol is BLM, also known as RECQL3, RECQ2
-
The gene locus is 15q26.1. The mRNA contains 4,437 base pairs.
-
The gene product is DNA helicase RecQ--like 3, one of 5 members of the RecQ helicase family. The normal protein functions as a caretaker tumor suppressor gene; it is essential for the maintenance of genome stability because it suppresses inappropriate recombination. BLM forms part of a multienzyme complex including topoisomerase III alpha (Top3a), replication protein A (RPA), and BLAP75/RMI1 to catalyze dissolution of double Holliday junctions at stalled replication forks. BLM interacts with DNA damage response proteins 53BP1, H2AX, FEN1, and colocalizes with the Fanconi anemia pathway protein FACND2. BLM complexes with TRF2 and has a role in telomere maintenance. Lack of BLM results in hyper-recombination and telomere association, to genomic instability and cancer predisposition.
-
Common mutations I are 2281del6/ins7 in Ashkenazim and 631delCAA in Japanese individuals.
Laboratory studies
A typical cytogenetic finding from cytogenic analysis in Bloom syndrome is the quadriradial configuration, which is produced by chromatid rearrangements. The 4-armed figure consists of 2 homologous chromosomes caused by chromosome breaks and rearrangements. Quadriradials also may be seen in some heterozygous males' sperm. Another cytogenetic abnormality observed in Bloom syndrome is a sharply increased SCE level.
Patients with Bloom syndrome have decreased amounts of circulating IgA or IgM and possibly IgG. DNA diagnostics may detect BLM gene mutations. In the United States, several centers offer direct DNA mutation analysis. A federally funded online directory of US genetics laboratories that offer DNA mutation analysis, protein analysis, or both is available at GeneTests. Israel has a center specializing in Bloom syndrome that offers testing. The American College of Obstetrics and Gynecology recommends that individuals of Ashkenazi Jewish descent be tested for 4 disorders; several laboratories offer simultaneous carrier screening for as many as 6 additional disorders, including Bloom syndrome (2281del6/ins7). [17]
Medical care
Because Bloom syndrome has no specific treatment, physicians must treat the symptoms and major conditions produced by the disorder.
Consultations
The following consultations are indicated:
-
Medical geneticist for genetic diagnosis and genetic counseling
-
Dermatologist for telangiectasias
-
Pediatric infectious disease specialist for antibiotics
-
Hematologist-oncologist to monitor and treat malignancies
-
Endocrinologist for short stature and management of diabetes mellitus
-
Ophthalmologist for retinal abnormalities
-
Pediatric gastroenterologist for feeding problems, decreased interest in feeding, and episodes of regurgitation and vomiting
-
Psychosocial specialist to provide support for social interactions at home and school because the small size of affected patients does not correlate with their intellectual and emotional maturity (Families may benefit from counseling regarding the challenge of providing long-term surveillance for a wide variety of cancers in these patients.)
Patient education
Advise patients that protective sunscreens and clothing help minimize damage caused by sunlight. Sun precautions should be started in infancy.
Advise patients to minimize exposures to possible mutagenic agents, such as UV radiation.
Special concerns
Prenatal diagnosis is possible by measuring the sharply increased SCE level. The technique has been used in amniocentesis and chorionic villi sampling.
If the 2 BLM mutations of an affected child or carrier parents are known, prenatal diagnosis for pregnancies at increased risk by analysis of DNA extracted from fetal cells obtained by amniocentesis or chorionic villus sampling (CVS) is possible.
Although few patients with Bloom syndrome have reproduced, patients should be advised of the chances of having a child affected with Bloom syndrome. Risk depends on factors such as the ethnicity of the spouse and whether the parents are consanguineous.
Hypersensitivity to both DNA-damaging chemicals and ionizing radiation necessitates modification of standard cancer treatment regimens (ie, reduction of both dosage and duration).
Medical/legal pitfalls
Pitfalls include the following:
-
Failure to counsel the parents of an affected child about recurrence risks in future pregnancies
-
Failure to advise a patient of the future risk of having children affected with this disorder
-
Because of the relatively increased carrier rate in Ashkenazi Jews, population screening for Bloom syndrome is available for individuals of reproductive age. [18] In population screening of people with Ashkenazi Jewish heritage, targeted mutation analysis for common 2281del6/ins7 allele.
-
Failure to consider Bloom syndrome in differential diagnosis of children with short stature before treatment with growth hormone therapy [19]
Fanconi Anemia
Fanconi anemia (FA), also known as Fanconi pancytopenia, pancytopenia dysmelia, congenital hypoplastic anemia, constitutional infantile panmyelopathy, or congenital pancytopenia, is a rare autosomal recessive disorder first described by Guido Fanconi in 1927. In 1964, Schroeder et al described the chromosomal instability as an increased number of chromosomal breaks, gaps, rearrangements, and endoreduplication in cells grown in vitro.
 A 3-year-old patient with Fanconi anemia. Note the multiple birth defects, including short stature, microcephaly, microphthalmia, epicanthal folds, dangling thumbs, site of ureteral reimplantation, congenital dislocated hips, and rocker bottom feet. (Alter BP, Young NS. The bone marrow failure syndromes. In: Nathan DG, Oski FA, eds. Hematology of Infancy and Childhood, 4th ed. Philadelphia, PA: WB Saunders, Inc, 1993: 216-316.)
A 3-year-old patient with Fanconi anemia. Note the multiple birth defects, including short stature, microcephaly, microphthalmia, epicanthal folds, dangling thumbs, site of ureteral reimplantation, congenital dislocated hips, and rocker bottom feet. (Alter BP, Young NS. The bone marrow failure syndromes. In: Nathan DG, Oski FA, eds. Hematology of Infancy and Childhood, 4th ed. Philadelphia, PA: WB Saunders, Inc, 1993: 216-316.)
Chromosomal interchanges result from double strand breaks in S phase and involve nonhomologous chromosomal regions. In patients with Fanconi anemia, the breaks and interchanges occur in more than 30% of the cells. Chromosomal breakage increases with exposure to mitomycin C (MMC), DEB, and cisplatin. Typically, no SCEs are observed.
The syndrome is characterized by pancytopenia, varied congenital musculoskeletal and urogenital anomalies, hyperpigmentation, or hypopigmentation, developmental delay, and increased susceptibility to leukemia and other malignancies. Prognosis is generally poor; however, some patients have a milder phenotype. One third to one fourth of patients have no physical abnormalities, some may have several cell lines with one being normal, and some have no bone marrow hypoplasia.
Pathophysiology
The disorder affects all bone marrow elements and is associated with cardiac, renal, GI, oral, ear, and limb malformations; dermal pigmentary changes; hypogonadism; and solid tumors.
Onset of pancytopenia and bone marrow hypoplasia occurs in preadolescence and may continue to worsen; it is present in about 90% patients in their forties. Risk factors for preadolescent bone marrow failure (BMF) are abnormal radii and a 5-item congenital abnormality score. The lowest risk group has an 18% risk of BMF, whereas the highest risk group has an 83% risk. Survivors are at a higher risk of developing myelodysplasia (MDS) that progresses to acute myeloid leukemia (AML) and to solid tumors.
Growth retardation may begin in utero and continue postnatally. Major anomalies occur, especially in the radii, thumbs, and kidneys. Others include small stature, small eyes, microcephalus, infantile facies, scoliosis, and, occasionally, hip dislocation. However, about one third of patients have no physical abnormalities; some do not have BMF.
Many endocrine disorders have been identified in Fanconi anemia and may contribute to the growth deficiency. [20] External ear anomalies may be associated with hearing disorders such as conductive hearing loss. Patchy brownish skin discoloration in patients is caused by melanin deposition (café-au-lait spots). This discoloration ranges in size, from small areas to large patches with diffuse boundaries. One fourth of patients with Fanconi anemia have mental deficiency.
Respiratory tract infections may occur frequently. Diagnosis is often difficult because of the variation in both severity and pattern of developmental anomalies, as well as the age of pancytopenia onset.
Of the several types of cancer that may occur, myelodysplasia leading to acute myeloid leukemia is the most common. Among patients with Fanconi anemia (and presumably among heterozygous relatives), 10-15% have increased risk for leukemia and squamous cell carcinoma. Squamous cell carcinoma of head and neck, mostly in oral cavity or tongue, aerodigestive, and anogenital tracts, occurs at 100 times the frequency of that in the healthy population. Patients have increased susceptibility to human papilloma virus (HPV)–induced squamous cell carcinoma. [21] Solid tumors include medulloblastoma, Wilms' tumor, and breast cancer; they may occur before other symptoms are present.
Patients with Fanconi anemia and biallelic FANCD1/BRCA2 mutations develop leukemia at a median age of 2.2 years, in contrast to 13.4 years in other patients with Fanconi anemia. In fact, the cumulative risk of leukemia for this complementation group is 79% by age 10 years. [22] These patients are also at increased risk to develop certain solid tumors, including brain tumors, hepatoblastoma, and Wilms tumor. [23] The probability of a brain tumor, the most common solid tumor, is 85% by age 9 years, and the risk of any malignancy by age 5.2 years is 97%. Liver tumors (hepatic adenomas, hepatocellular carcinomas, and focal nodular hyperplasia) tend to occur in patients undergoing prolonged androgen therapy (introduced in 1959); these tumors may regress if androgen therapy is stopped. In some patients, a reversion mutation may lead to normal blood; these patients occasionally present with squamous cell carcinoma of the head and neck later in life.
About 84% of patients with Fanconi anemia and squamous cell carcinoma had human papillomavirus DNA in the tumors, compared with 36% of controls; the greater risk of patients may be due to a polymorphism in the tumor protein 53 gene, TP53. Cells of patients with Fanconi anemia are also transformed by simian virus more readily than the cells of unaffected individuals.
Frequency
The incidence is 1 per 100,000 live births. The overall prevalence of Fanconi anemia is estimated at 1 case per 360,000 people, with a resulting carrier frequency of 1 per 300 individuals. An unusually high prevalence of 1 case per 22,000 people, with a carrier frequency of 1 per 77 people, has been reported in white Afrikaans-speaking South Africans. The high incidence is thought to be the result of a founder effect because other South African populations have lower prevalences. Fewer than 5% of families with Fanconi anemia within the family have BRCA2 mutations.
Mortality/morbidity
Before therapy with androgens, survival following diagnosis of pancytopenia usually was 2 years. In a sample of 25 black South African children, the mean age at death from leukemia was 9.8 years, and death occurred about 2.3 years after diagnosis. According to the International Fanconi Anemia Registry, 73% of patients with Fanconi anemia develop overt bone marrow disease by age 10 years; subsequent median survival time is 7 years. According to a large International Bone Marrow Transplant Registry, 2-year survival probabilities were 66% after human leukocyte antigen (HLA)–matched sibling hematopoietic stem cell transplantation (HSCT) and 29% after unmatched donor HSCT.
Severe graft versus host disease (GVHD) decreased significantly; however, patients had additional lethal events related to head and neck carcinomas starting 5 years after transplantation and patients usually still develop myelodysplasia, acute myeloid leukemia, or solid tumors. [24]
Physical abnormalities associated with Fanconi anemia are generally not lethal.
Sex
Males and females appear to be affected in equal numbers for the autosomal recessive forms of Fanconi anemia. However, about 32% of males have abnormal genitalia compared with 3% of females. [25] For the X-linked complementation group of Fanconi anemia (FANCB), males are affected and females are unaffected carriers.
Age
Short stature often begins prenatally. The average age of onset of anemia is about 8 years but widely varies and occurs slightly earlier in males. Recurring infection usually appears in children aged 5-10 years. An early onset of malignancy occurs in 10-15% of affected patients.
Clinical outline
Fanconi anemia may be the most common chromosomal breakage syndrome, yet it is quite rare. The disorder's clinical picture is complicated by the multiple malformations associated with Fanconi anemia and the number of genetic complementation groups that cause Fanconi anemia. Various combinations of anomalies may be present in different patients. In addition, the severity of a malformation may be quite variable. When Fanconi anemia is suspected, despite mild features, examination of the cell cycle may be useful to determine if the gap 2 (G2) phase is prolonged.
At birth, the most common features of patients with Fanconi anemia are short stature and rudimentary or absent thumbs. Radial ray defects range from fingerlike thumbs to hypoplasia of the thumbs to complete radial aplasia. Among patients listed in the International Fanconi Anemia Registry, 60-75% have congenital malformations. Only 28% of the patients with congenital malformations were diagnosed with Fanconi anemia before the appearance of hematologic manifestations (evidence of the difficulty in diagnosing Fanconi anemia). Although the average age of onset of hematologic symptoms is about 8 years, in some people, these symptoms do not appear until postadolescence. The pancytopenia, however, is usually progressive, and anemia is often a cause of death in the young.
Hyperpigmentation and hypopigmentation of the skin or café-au-lait spots is also a common and useful clinical feature. The brown-pigmented areas also increase with age.
Patients with Fanconi anemia may have urogenital system abnormalities and should be evaluated for possible renal abnormalities. Anomalies include hydronephrosis, duplication of the ureter, and renal dysplasia.
Patients with Fanconi anemia are predisposed to leukemia and other malignancies. A somewhat increased risk for cancer is also noted in heterozygotes.
Recent studies suggest that approximately 5% of patients with Fanconi anemia also have the VACTERL phenotype, including radial ray anomalies. [26] The VACTERL with hydrocephalus (VACTERL-H) phenotype is exhibited by some patients with X-linked Fanconi anemia (complementation group FANCB). [27]
Other clinical features of patients with Fanconi anemia include asymmetric, bilateral conductive hearing loss that is more severe at lower frequencies and structural anomalies of the external ear. [28]
Causes
Cells have deficient ability to excise UV-induced pyrimidine dimers from the cellular DNA; they are sensitive to small concentrations of DNA crosslinking agents or lesions arising from oxidative damage. The defect may be in any of the proteins involved in DNA interstrand crosslink repair; it leads to double-strand breaks in the S phase of the cell cycle and accumulation of cells in G2.
Genetic causes can be summarized as follows:
-
The mutations causing Fanconi anemia are heterogeneic and exhibit an autosomal recessive mode of inheritance, with the exception of FANCB mutations, which are inherited in an X-linked manner. Currently, 15 complementation groups have been identified; the locations of all genes are known (see the table below).
-
Consanguinity is increased, and siblings are affected.
-
Some phenotype-genotype correlations occur: Some mutations cause mild disease; others cause severe disease.
-
Subtyping is recommended in newly diagnosed Fanconi anemia patients because of the clinical variability among complementation groups. In particular, patients with FANCA tend to have milder disease with later onset of bone marrow failure, patients with FANCC and FANCG mutations tend to have more severe disease necessitating earlier intervention, and FANCD1 patients have earlier onset and increased incidence of leukemia and solid tumors. [29]
Table 3. Gene Symbols and Synonyms With Location, Population, and Mutation (Open Table in a new window)
Gene |
Locus |
Population |
Mutation |
Comments |
FANCA, 60-70% of mutations |
16q24.3 |
Northern European Israeli Arab |
3788-3790del 1115-1118del exons6-31del IVS42-2A>C |
Nuclear core complex protein |
FANCB, 2% of mutations |
Xp22.31 |
... |
1838insT 10693del3314 1650delT 811insT |
Nuclear core complex protein, X-linked inheritance, X-inactivation skewed towards mutant allele in female carriers, some cases present as VACTERL-H |
FANCC, 14% of mutations |
9q22.3 |
Ashkenazim Japanese Northern European Northern European Southern Italian |
IVS4+4A>T R548X, L554P, R185X, 322delG, Q13X |
Nuclear core complex protein, milder phenotype in Japanese, birth defects and earlier hematologic abnormality, congenital abnormality and later bone marrow failure |
FANCD1/BRCA2 – ~3% of mutations |
13q12.3 |
Ashkenazim Ashkenazi/ Lithuanian Latin American African American |
7691insAT 9900insA 6174delT/ C3069X 6174delT/ 886delGT I2490T/ 5301insA Q3066X/ E1308X |
Early-onset leukemia and solid tumors (breast cancer, brain tumors, medulloblastoma) |
FANCD2, 3% of mutations |
3p25.3 |
... |
... |
Frequent malformations, earlier and more severe hematologic manifestations |
FANCE, 3% of mutations |
6p22-p21 |
... |
... |
Nuclear core complex protein |
FANCF, 2% of mutations |
11p15 |
... |
... |
Nuclear core complex protein |
FANCG/XRCC9, 10% of mutations |
9p13 |
Korean/Japanese Brazilian French Canadian Northern European Israeli Arab |
IVS3+1G>C IVS8-2A>G IVS11+1G>C 1184-1194del 1794-1803del IVS4+3A>G |
Nuclear core complex protein |
FANCH=FANCA |
... |
... |
... |
... |
FANCI, 1% of mutations |
15q25-q26 |
... |
... |
Undergoes ubiquination and interacts with FANCD2 |
FANCJ/BRIP1/BACH1, 2% of mutations |
17q23 |
Many Argentinian |
R798X Y800X W647C, R707C |
Helicase that interacts with FANCD2 and BRCA1 |
FANCL/FAAP43,< 1% of mutations |
2p16.1 |
... |
... |
Ubiquitin protein ligase, nuclear core complex protein |
FANCM/KIAA1596,< 1% of mutations |
14q21.3 |
... |
... |
ATP-dependent RNA helicase, nuclear core complex protein |
FANCN/PALB2< 1% of mutations |
16p12 |
... |
... |
Early-onset leukemia and solid tumors, partner and localizer of BRCA2 |
FANCO/RAD51C< 1% of mutations |
17q22 |
... |
... |
DNA repair protein |
FANCP/SLX4< 1% of mutations |
16p13.3 |
... |
... |
Structure-specific endonuclease subunit |
Upon DNA damage during replication or during the S phase of the cell cycle, FANCA, FANCB, FANCC, FANCE, FANCF, FANCG, FANCL, and FANCM transcripts assemble into a nuclear core complex that ubiquinates FANCD2 and FANCI, which then interact with FANCD1/BRCA2, RAD51C, FANCJ/BRIP1, BRCA1, BLM, ATM, ATR, NBS1,SLX4, and PALB2 for DNA repair and genomic stability.
Laboratory studies
Cytogenetic analysis, analysis of blood, bone marrow, skin cells, amniocytes, or chorionic villi, is available in 5 laboratories (see GeneTests). Patients with Fanconi anemia have increased chromosomal breakage after culturing with a DNA interstrand crosslinking agent such as DEB or mitomycin C (MMC), cisplatin, or photoactivated psoralens, which arrest cells in late S phase of the cell cycle. This increased breakage occurs even in the absence of other symptoms. However, the test does not identify heterozygotes (carriers); unaffected siblings of a patient with Fanconi anemia with a normal chromosomal breakage have a two thirds risk of being carriers. The test does not identify female carriers of X-linked Fanconi anemia due to skewed X-inactivation of the mutated FANCB allele in most cells.
Common cytogenetic abnormalities are monosomy 7; deletions of the long arms of chromosomes 5, 7, and 20 (5q−, 7q−, 20q−); trisomy 8; and translocations and rearrangements of chromosomes 1 and 3. Note that patients may have 2 or more cell lines, one of which may be normal. The normal cell line is thought to arise from back mutation, gene conversion, and selective loss of the abnormal cell line. Also found were revertant cells in mature myeloid cells but not in peripheral blood cells. If one cell type is normal despite Fanconi anemia symptoms, testing should be done using another cell type.
Targeted mutation analysis is widely available for the common Ashkenazi Jewish FANCC mutation (c.345+4A).
DNA diagnostics should be used to identify the specific mutation. It can be used to confirm the diagnosis or for carrier detection, as well as for prenatal diagnosis and preimplantation diagnosis. Clinical testing, including mutation analysis and gene sequencing, is available for mutations in FANCA, FANCB, FANCC, FANCD1/BRCA2, FANCE, FANCF, FANCG, FANCJ, FANCL, FANCM, and FANCN. In the United States, several centers offer clinical testing; a federally funded online directory of US genetics laboratories is available at GeneTests. Mutation analysis of FANCD2, FANCI, RAD51C, and SLX4 may be available in research laboratories.
In families with X-linked Fanconi anemia, testing of suspected female carriers depends upon identification of the specific familial FANCB mutation.
Single-parameter flow cytometry can be used to detect an abnormally large proportion of cells in the G2 phase of the cell cycle, which results from the arrest of cells in late S phase.
Other laboratory findings may include RBC macrocytosis, elevated fetal hemoglobin, and erythrocyte i antigen, which may precede the onset of anemia but has no prognostic significance. Erythropoietin concentration may be normal or elevated. [25]
Prenatal testing and preimplantation genetic diagnosis may be available for families in which the disease causing mutations have been identified.
Chromosomal breakage studies should be considered in the evaluation of patients with features of the VACTERL association, with and without hydrocephalus, especially in cases with radial-ray anomalies and Fanconi anemia–associated manifestations, such as skin pigmentation abnormalities, growth retardation, microcephaly, and microphthalmia.
Medical care
Hearing should be formally evaluated. The kidneys and urinary tract should be examined via ultrasonography. Development, especially of preschool and school-aged children should be assessed.
Androgen therapy improves the blood counts (red cell, white cell, and platelets) in half of Fanconi anemia patients treated. The standard recommended androgen is oxymetholone at a starting dose of 2-5 mg/kg/d given orally. [25] Side effects are elevated liver enzymes, cholestasis, peliosis hepatis, and liver tumors.
Bone marrow transplantation of hematopoietic stem cells (HSC) can be curative for hematologic symptoms. Due to the sensitivity of patients with Fanconi anemia to chemotherapy and radiation, reduced doses are administered. However, patients with FANCF - or FANCB -mutated tumors might be effectively treated with crosslinking chemotherapeutic agents, such as cisplatin. Bone marrow transplantation for myelodysplasia and acute myeloid leukemia can be performed at centers experienced in the treatment of Fanconi anemia. Treated patients continue to have a higher risk of solid tumors than the general population.
Patients with Fanconi anemia who receive HSC transplant may develop late onset of isolated thrombocytopenia resulting in chimerism and complete graft failure. The absence of fludarabine in the preparative regimen has been reported as a potential risk factor. [30]
A mathematical decision model developed by Khan et al indicated that preemptive bone marrow transplantation in young patients with Fanconi anemia increases expected mean event-free survival, with the procedure in these patients associated with little carcinogenicity and only a small risk of transplantation-related mortality. In adults, however, the model predicted that preemptive bone marrow transplantation will actually reduce expected event-free survival, since the risk of posttransplantation solid tumor development is higher in these patients. [31]
Transfusions of red cells or platelets should be avoided for patients who are candidates for hematopoietic stem cell transplantation to avoid sensitization of the patient. If blood transfusions are done, blood products should be leukodepleted and irradiated. [25]
One patient was successfully treated with antilymphocyte globulin and subsequent donor lymphocyte infusion.
Umbilical cord blood hematopoietic stem cell transplantation from an HLA-identical and unaffected sibling of the patient cured the patient's bone marrow failure without subsequent GVHD; the technology involved preimplantation genetic diagnosis and in vitro fertilization. [32] Recent studies demonstrate that fludarabine containing a nonmyeloablative conditioning regimen avoids the toxicity of high dose irradiation. [33]
Malignancy surveillance to detect and remove tumors at an early stage; however, radiographic tests should be avoided. Examinations should include dental and oropharyngeal check-ups, annual esophageal endoscopy and annual gynecological examination, Papanicolaou test, and rectal examination.
Gene therapy studies are underway, as are clinical trials of improved treatment for malignancies associated with Fanconi anemia. Searching for ongoing clinical trials is advisable.
Consultations
Refer affected individuals to an endocrinologist. Consult a medical geneticist for diagnosis and genetic counseling. All genes identified to date, except FANCB, are inherited in an autosomal recessive manner; FANCB is located on the X chromosome and inherited in an X-linked recessive manner. In females, X inactivation appears to be selective for the abnormal allele. Consult an ophthalmologist.
Consult a dentist to check for oral lesions, gingival and periodontal status, and tooth decay. An increased prevalence of periodontal disease in patients with Fanconi anemia may be due to anemia, leucopenia, oxygen radicals, as well as to immunosuppressive medication.
Consult a hematologist to monitor bone marrow development, blood counts, and blood chemistries. Patients and their parents and siblings should be HLA-typed for possible bone marrow transplantation. An annual bone marrow biopsy is recommended and should include cytogenetic analysis.
Special concerns
Because early detection is the best hope for the best outcome for treatment of Fanconi anemia, chromosome breakage studies should be considered in the evaluation of any infant born with any type of radial ray anomaly.
The DEB-induced chromosomal breakage test can be used to screen for Fanconi anemia in amniotic fluid or chorionic villus cells. In a study reported by Auerbach et al, of the 30 at-risk fetuses that were involved, 7 of the fetuses were diagnosed as affected, of which, 2 were carried to term and were affected clinically. [34] The 23 otherwise healthy fetuses were born with no evidence of Fanconi anemia.
DNA banking is advisable and provides a means for testing when all the genes involved in the disorders have been identified and sequenced and methodologies to detect the mutations are established.
Although reduced fertility is one clinical manifestation of Fanconi anemia, the rate of successful pregnancy in nontransplanted Fanconi anemia patients who reach adult age has been estimated at 15%. [35] Although females with Fanconi anemia may develop secondary infertility after hematopoietic stem cell (HSC) transplantation using an attenuated conditioning regime of low-dose cyclophosphamide, recovery of normal ovarian function and successful pregnancy have been achieved in a small minority. [36] Patients should be counseled before HSC transplantation regarding the risk of reduced fertility and other methods of procreation, including cryopreservation of ovarian tissues before transplantation and new techniques of in vitro fertilization.
Medical/legal pitfalls
Pitfalls include the following:
-
Failure to perform chromosomal breakage studies on all babies and children with the VACTERL phenotype, with and without hydrocephalus, especially in cases with radial-ray abnormalities and other manifestations such as skin pigmentation abnormalities, growth retardation, microcephaly, and microphthalmia.
-
Failure to advise a patient of the future risk of having children affected with this disorder
-
Failure to counsel the parents of an affected child about recurrence risks in future pregnancies
-
Failure to consider chromosomal breakage studies or molecular genetic testing (if the specific familial mutation(s) is known) on all siblings of an affected individual
-
Failure to advise families of the technology of HSC transplantation with umbilical stem cells obtained from an unaffected embryo that is HLA-identical to the patient [32]
-
Failure to counsel a female patient considering stem cell transplantation regarding the risk of reduced fertility and other available methods of procreation
Animal studies
Human bone marrow cells containing retrovirally transferred FANCA -cDNA were cultured in conditions that limited oxidative stress and transplanted into nonobese diabetic (NOD)/severe combined immunodeficiency mice; the FANCA transgene could be detected 6 months later.
Knockout mice were constructed for research of FANCA, FANCC, FANCG, and FANCD2, as well as double knockout for FANCA and FANCC. No developmental abnormalities except for a decrease in the number of germ cells and no spontaneous hematological abnormalities were observed, only FANCD2 mice had increased malignancies. Homozygous deletion of FANCD1/BRCA2 or PALB2 led to embryonic death in mice. Mice with cells whose FANCA mutation had been corrected survived 6 months.
Knockout chicken for FANCJ/BRIP1/BACH1 revealed that the gene product is a DEAH helicase that interacts with BRCA1.
Resources
Children's Hospital Boston
Bone Marrow Failure Program
300 Longwood Avenue
Boston, MA 02115
Tel: 617-355-8246
Fanconi Anemia Comprehensive Care Center
Cincinnati Children's Hospital Medical Center
3333 Burnet Avenue
Mail Location 2022
Cincinnati, OH 45229-3039
Tel: 800-344-2462 Ext 3218; 513-636-3218
Fanconi Anemia Research Fund, Inc
1801 Willamette Street, Suite 200
Eugene, OR 97401
Tel: 800-828-4891; 541-687-4658
Fax: 541-687-0548
Email: info@fanconi.org
Xeroderma Pigmentosum
Moriz Kaposi first described the disorder in 1876 and named it xeroderma pigmentosum (XP) in 1882. Xeroderma pigmentosum is usually included with the chromosomal breakage syndromes because it involves defects in the nucleotide excision repair (NER) of DNA, although no abnormality is evident at the cytogenetic level. The disorder is extremely rare.
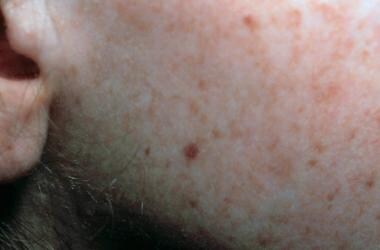 Face of a toddler with xeroderma pigmentosum, representative of an early stage of the disease. Note the freckling and the scaling. Courtesy of Neil S. Prose, MD, Duke University Medical Center, Durham, North Carolina.
Face of a toddler with xeroderma pigmentosum, representative of an early stage of the disease. Note the freckling and the scaling. Courtesy of Neil S. Prose, MD, Duke University Medical Center, Durham, North Carolina.
Xeroderma pigmentosum is a heterogeneous group of autosomal recessive conditions caused by mutations in at least 10 genes and 9 complementation groups.
Patients affected by this disorder have extreme photosensitivity and photophobia and develop freckling and premalignant and malignant skin lesions arising in keratinocytes soon after even the briefest exposure to sunlight. Patients are also hypersensitive to environmental mutagens such as cigarette smoke and probably to the widely-used agricultural insecticide, diazinon.
Patients usually develop skin cancers in areas exposed to UV light.
Pathophysiology
The underlying biochemical defect in patients with xeroderma pigmentosum is a malfunction of an enzyme involved in nucleotide excision repair. Defective DNA repair occurs after exposure to UV light, usually in the range of 290-320 nm, or chemical carcinogens. The level at which the repair defect occurs depends on which of the genetic complementation groups is involved. Patients in the xeroderma pigmentosum variant complementation group (also known as the pigmented xerodermoid) have normal nucleotide excision repair post-UV exposure but defective postreplication repair. Except for patients in the XPG complementation group, patients are not usually hypersensitive to X irradiation.
Many clinical features are caused by extreme sun sensitivity of the skin and cornea; severe changes appear in children younger than 3 years and become progressively more severe. Early symptoms are diffuse erythema, freckles of various sizes and color, and telangiectasias on exposed areas of the skin as well as dryness, scaling, atrophy, and numerous actinic keratoses on the face and actinic cheilitis on the lips. Telangiectasias may appear even in buccal mucosa and on unexposed skin. Patients in the XPE complementation group generally have mild skin abnormalities.
The risk of cancer in patients with early xeroderma pigmentosum onset is 1000 times higher by age 20 years than that for the general population. Basal cell carcinoma, squamous cell carcinoma, malignant melanoma, and fibrosarcoma occur in large numbers by age 4-5 years, as do keratoacanthomas and benign and malignant tumors of ectodermal and mesodermal origin. (In unaffected individuals, UVB radiation may lead to nonmelanoma skin cancer but not to cutaneous malignant melanoma.) Other tumors that occur at a 10-fold to 20-fold increased frequency are squamous cell carcinoma in the oral cavity (particularly the tip of the tongue) and tumors in breast, lung, pancreas, stomach, kidney, and testicles. Sarcomas and adenocarcinomas are also more frequent, and some patients have leukemia. Although heterozygotes (carriers) are not affected, some may have an increased risk of skin or lung cancer or an altered response to chemotherapeutic agents.
Ocular abnormalities are nearly as common as those of skin. They tend to start with photophobia and conjunctivitis; eyelid lentigines develop in the anterior portion of the eyes in the first decade of life and progress to severe inflammation of the cornea, opacification, vascularization, and malignant melanoma. The eyelids may turn outward, and repeated inflammation and infection of the conjunctiva may lead to scarring. The skin of the eyelids may atrophy and eventually lead to loss of the lids. Patients may develop epithelioma, basal cell carcinoma, squamous cell carcinoma, fibrosarcoma, and melanoma involving the eyes; these may be more severe in dark-skinned individuals.
The condition may lead to stunted growth, blindness, sensorineural deafness, and other neurological deficits. Some patients exhibit microcephaly, spasticity, and ataxia. Neurologic deficits are not preventable. The different complementation groups have different degrees of neurological effects; these tend to worsen slowly. They are more common in patients in complementation groups XPA and XPD. Symptoms include afebrile or more rarely febrile convulsions, epilepsy, microcephaly, progressive intellectual impairment, hearing loss starting with loss of high frequency tones, spasticity, hyporeflexia or areflexia, ataxia, and intellectual disability.
Patients in the XPV/POLH complementation group do not have neurologic abnormalities. Patients in complementation groups XPC, XPE, XPF, or XPG generally do not have neurological abnormalities; however, those in the XPG group may present with xeroderma pigmentosum/Cockayne syndrome (XP/CS) complex. In addition, mutations in xeroderma pigmentosum genes (ERCC3, ERCC4, ERCC1) have been described in patients with other conditions including trichothiodystrophy, xeroderma pigmentosum/trichothiodystrophy, cerebral-ocular-facial-skeletal syndrome (also known as Pena-Shokeir syndrome type II).
Patients with xeroderma pigmentosum/Cockayne syndrome complex have facial freckling, early onset and more frequent skin cancers, intellectual disability, spasticity, dwarfism, and hypogonadism but no skeletal dysplasia.
Note that a diagnosis of De Sanctis-Cacchione syndrome should be made only for patients with xeroderma pigmentosum who have severe neurological degeneration, short stature, and immature sexual development. De Sanctis-Cacchione syndrome is a rare clinical entity that can be displayed by patients with different XP complementation groups, especially XPA.
Trichothiodystrophy without xeroderma pigmentosum (TTD) presents with variable symptoms including hypersensitivity to UV rays, ichthyosis (rough, thick, and scaly skin), brittle hair, intellectual impairment, short stature, microcephaly, protruding ears, micrognathia, and decreased fertility, but patients with trichothiodystrophy do not develop skin neoplasias. However, patients with TTD have a 20-fold increased risk of death before age 10 years, primarily from infections. [37] Mutations in XPD/ERCC2 and ERCC3 have been demonstrated in some patients with TTD.
Patients with XP/trichothiodystrophy syndrome present with clinical features of TTD and XP, including an increased frequency of skin cancers, and specific mutations in the XPD/ERCC2 gene.
Cerebral-ocular-facial-skeletal (COFS) syndrome without xeroderma pigmentosum is a progressive neurological disorder associated with microcephaly with intracranial calcifications, growth retardation, microcornea, cataracts, optic atrophy, and joint contractures, but patients do not develop skin neoplasias. Mutations in XPD/ERCC2 have been demonstrated in some patients with COFS; only one affected individual is reported to have ERCC1 mutations.
Some patients of the XPA complementation group develop a harsh, high-pitched respiratory sound in their third decade of life, which is caused by laryngeal dystonia.
UVB irradiation has an immunosuppressive effect, and patients are susceptible to infection.
Frequency
Xeroderma pigmentosum occurs at a frequency of 1 case per million people in the United States. A higher prevalence of xeroderma pigmentosum is found in Japan, with 1 case per 22,000 people. [38] The prevalence is increased in North Africa (Morocco, Algeria, Tunisia, Libya, Egypt) and in the Middle East (Israel, Syria, Turkey), particularly in areas where consanguinity is common. [39]
Morbidity/mortality
Without early diagnosis and cancer screening, many patients with this condition die during childhood. Fewer than 40% survive beyond age 20 years; few survive to age 30 years. Patients with milder phenotypes may live beyond middle age. Children with skin cancer tend to have a better prognosis than do adults with skin cancer.
Symptoms are progressive in patients with neurologic involvement. Frequent surgeries for facial cancers may also have a severe psychological impact.
Sex
Both sexes are affected equally.
Age
Xeroderma pigmentosum may be diagnosed during the first year of life; median age of diagnosis is age 1-2 years. Infants may display photophobia and develop persistent erythema following minimal exposure to sunlight. Squamous cell skin cancer may develop in children younger than 8 years; median onset of nonmelanoma skin cancers is in children younger than 10 years. The degree of protection from sunlight contributes to varying ages of onset. Approximately 25% of affected patients have neurological abnormalities that may present in infancy but are delayed in some individuals until the second decade or later. [40] Patients in the XPV complementation group may not develop symptoms until the third decade of life.
Clinical outline
Symptom severity depends on the specific xeroderma pigmentosum mutation and the child's history of exposure to sunlight. During infancy, acute sensitivity to sunlight manifests as a severe sunburn erythema. During childhood, pigmented macules and telangiectasias develop; their distribution relates to the skin areas exposed to sunlight. Skin carcinomas and malignant melanomas may also develop during this period. Ophthalmologic abnormalities are limited to the anterior, UV-exposed portion of the eyes. [41] Effective xeroderma pigmentosum management requires early diagnosis. Sun avoidance and protection minimize many of the severe effects of xeroderma pigmentosum and allow patients to reach adulthood.
Causes
See the list below:
-
Defective nucleotide excision repair (NER) of pyrimidine dimers upon UV exposure
-
Defective postreplication repair in the xeroderma pigmentosum variant complementation group
Genetic causes can be summarized as follows:
-
Autosomal recessive mode of inheritance
-
Frequently, parental consanguinity
-
Subgroups XPA-XPG and XPV
XPA – Accounts for 25% of mutations
9q22.3
DNA-repair protein, recruited to photolesion by tumor suppressor p33ING2
Most frequent subgroup among Japanese individuals; most common mutation is IVS3AS,G-C, a splicing mutation in exon 3
Causes skin defects and mild to severe neurologic symptoms
XPB/ERCC3 - Rare
2q21
Helicase subunit
Can cause xeroderma pigmentosum/Cockayne syndrome complex, trichothiodystrophy, and xeroderma pigmentosum with mild neurologic abnormalities
XPC – Accounts for 25% of mutations
3p25
DNA-repair protein
Most common subgroup in the United States
Causes skin defects and can cause neurological symptoms; in German Caucasian patients, the XPC haplotype IVS9 PAT+, IVS11-6A, and exon 15 2920C may increase risk to cutaneous melanoma
XPD/ERCC2 – Accounts for 15% of mutations
19q13.2-q13.3
Helicase subunit
Involved in unwinding a damaged DNA region
Can cause xeroderma pigmentosum with or without neurologic abnormalities, xeroderma pigmentosum/Cockayne syndrome complex, trichothiodystrophy, xeroderma pigmentosum/trichothiodystrophy, cerebral-ocular-facial-skeletal syndrome
XPE/DDB2 - Rare
11p12-p11
DNA damage binding protein; defective protein cannot regulate the tumor suppressor p53 (TP53)–mediated responses after UV irradiation, which results in decreased p53-mediated apoptosis
XPF/ERCC4 – Accounts for 6% of mutations
16p13.3-p13.13
5' endonuclease
Found in Japanese
XPG/ERCC5 – Accounts for 6% of mutations
13q33
3' endonuclease
Missense mutations lead to xeroderma pigmentosum without neurological symptoms while mutations that result in truncated proteins lead to xeroderma pigmentosum/Cockayne syndrome complex
Mutations in subgroups XPA-XPG cause failure to repair excisions.
XPV/POLH – Accounts for 21% of mutations
6p21.1-p12
Polymerase eta; in normal form is an error-prone translesion DNA synthesis polymerase that can bypass UV-induced cyclobutane pyrimidine dimers, has a role in the DNA damage checkpoint involving p53, and is involved in immunoglobulin-variable hypermutation
XPV/POLH- deficient cells are hypersensitive to the chemotherapeutic platinum-based drugs cisplatin, carboplatin, and oxaliplatin.
Mutations lack error-free DNA polymerase eta.
Reported in United States, Europe, and Japan
Causes mild to severe skin symptoms but no neurologic symptoms
XPB and XPD are also involved in regulation of the basal rate of RNA synthesis of active genes.
ERCC1
19q13.32
Described in one affected individual with severe clinical features consistent with a diagnosis of COFS and demonstrating moderate hypersensitivity to UV rays and mitomycin C. [42]
The normal gene products are part of the nucleotide excision repair (NER) system that recognizes DNA errors, particularly UV-induced pyrimidine dimers, during replication as well as during transcription; xeroderma pigmentosum is caused by defects in any of 8 genes identified so far that prevent nucleotide excision repair during replication. Some proteins, including those for XPC and probably XPE/DDB2 bind to the damaged site; XPB and XPD partially unwind the DNA in the damaged region; XPA may function in conjunction with proteins for RPA, TFIIH, and ERCC1.XPF in a complex with ERCC1 makes a single-strand nick at the 5' side of the damaged DNA, while XPG makes a nick on the 3' side. The resulting gap is filled by DNA polymerase involving proliferating cell nuclear antigen; DNA ligase I then joins the cut ends.
Laboratory studies
See the list below:
-
Cytogenetic analysis is available in clinical laboratories and can be used to assess chromosomal breakage, which can also be used for prenatal diagnosis on amniocytes from at-risk fetuses.
-
Unscheduled DNA synthesis measures DNA repair synthesis after excision of a DNA segment containing damage caused by UV or chemical agents and replacement with normal DNA. The test can be performed on cultured skin fibroblasts and amniotic fluid cells. The test may be available in a research laboratory.
-
Host-cell reactivation is used to determine the xeroderma pigmentosum complementation group. This test may be available in a research laboratory.
-
Hypersensitivity testing to UV radiation may be available in a research laboratory.
-
Molecular genetic testing by gene sequencing is clinically available for some complementation groups (XPA, XPC); testing on a research basis may be available for other XP genes (see GeneTests). When a patient's mutations are known, the parents are obligate carriers (heterozygotes) and unaffected siblings of the patients can be tested for carrier status; prenatal diagnosis may also be available. In Japan, 90% of patients with xeroderma pigmentosum have the same single-base substitution mutation in XPA. A rapid test for this mutation can detect homozygotes (affected) and heterozygotes and can also be used for prenatal diagnosis.
-
Prenatal diagnosis for XP in at-risk pregnancies can be made by demonstrating reduced UV-induced DNA repair synthesis in cultured chorionic villus cells. [43]
Medical care
Individuals with clinical findings suggestive for XP, such as severe burning on UV exposure or frecklelike pigmentary abnormalities before age 2 years, should immediately begin UV-protective measures before a diagnosis is established.
A detailed family history, including possible consanguinity, should be obtained.
Skin, especially in sun-exposed areas, should be examined clinically every 3-6 months. Examine lips for inflammation and mucous membranes for permanent dilation of blood vessels that may lead to neoplasias. Dermoscopic examination may facilitate the discrimination of melanomas, basal cell carcinoma, and dysplastic nevi from other pigmented skin lesions. Document symptoms with color photographs and a ruler to evaluate progression of the disease.
Examine eyelids, including the underside, as well as anterior portions of the eyeball. Perform a test for dry eyes. Patients should wear UV-absorbing sunglasses and, if eyes are dry, use gels or solutions for this condition. Corneal transplants may restore vision.
The most effective early treatment is careful and routine sun avoidance and the use of protective clothing and sunscreens. Patients must wear UV-protective glasses with side-shields. Sunscreens with high sun-protective factor (SPF) should be used even in winter months and in dusk and dawn. Physical sunscreens scatter and reflect radiation; they block UV, infrared rays, and visible light. Chemical sunscreens absorb UV rays. A component of some sunscreens, p-aminobenzoic acid (PABA), can cause allergic reactions. Broad-spectrum chemical sunscreens block UVA and UVB, and many are water-resistant.
Since patients with xeroderma pigmentosum are hypersensitive to UVA, UVB, and UVC rays, a light meter should be used to detect and eliminate high levels of environmental UV in places frequented by the patient; halogen lamps emit high levels of UV.
Chemical therapies are available. In some patients, skin cancers regressed with the use of imiquimod cream. Retinoids decrease the occurrence of skin cancer in patients with xeroderma pigmentosum, but they have deleterious side effects and must be avoided during pregnancy and breastfeeding. Coadministration with vitamin A, tetracyclines, benzoyl peroxide, resorcinol, certain soaps, or alcohol-containing products results in toxicity or other adverse events. Chemical therapy with 5-fluorouracil in combination with oral acitretin resolved premalignant actinic keratoses within 6 months. Topical application of DNA repair enzymes to UV-damaged skin decreased the appearance of actinic keratoses and basal cell carcinomas in one year. Note that XPV/POLH- deficient cells are resistant to platinum-based chemotherapeutic drugs cisplatin, carboplatin, and oxaliplatin.
Tumors may be treated with X irradiation except in patients of the XPG complementation group.
Other treatments are liquid nitrogen, therapeutic dermatome shaving, dermabrasion (while monitoring systemic complications that may lead to cardiac arrhythmias or sudden death), electrodesiccation and curettage, surgical excision, or chemosurgery. Ocular tumors should be removed surgically. Surgical excision should remove the tumor completely but without removing undamaged skin. Skin grafting from unaffected areas can be performed.
Gene therapy with retrovirus vectors may be possible for patients of the XPC complementation group. Treatment with a bacterial enzyme is in progress. It is advisable to search for ongoing clinical trials.
Head circumference should be measured. Neurological examinations should be performed routinely. Symptoms may not be present in young children, but when they appear they may worsen. Deep tendon reflex testing should be performed, as well as a hearing test (audiometry) to detect early high-tone hearing loss. If neurological abnormalities are detected, CT scan and MRI of the brain may show enlarged ventricles and thinning of the cortex. Nerve conduction velocities and an electromyogram may reveal neuropathy.
Avoidance of solar UV radiation may cause vitamin D deficiency. Serum 25-hydroxyvitamin D levels should be measured and, if suboptimal, vitamin D supplementation should begin. [44]
Consultations
Referral to appropriate specialists is crucial. These may include a neurologist, ophthalmologist, dermatologist, oncologist, and others, as necessary.
Refer affected individuals to a medical geneticist for diagnosis and genetic counseling.
Refer patients for appropriate surgical procedures when necessary.
Special concerns
Because xeroderma pigmentosum is highly heterogeneous genetically, attempt to determine the specific gene mutation and complementation group involved. Accurate determination helps give patients and families more precise information about the patient's prognosis and the risk with future pregnancies.
Animal models
Research with knockout XPD/ERCC2 mice revealed that such mice do not survive; however, XPA and XPC mice are viable. XPV/POLH -deficient mice are viable, fertile, and show no defects in their first year of life, but all develop skin tumors after UV-irradiation, while the wild-type litter mates did not.
Recombinant adenovirus carrying human XPA was used for in vivo gene therapy in UVB-irradiated skin of xeroderma pigmentosum–mutant mice; XPA was expressed in keratinocytes and prevented deleterious effects in the skin, including late development of squamous cell carcinoma.
Medical/legal pitfalls
Failure to advice a patient of the future risk of having children affected with this disorder is a medicolegal issue.
Failure to counsel the parents of an affected child about recurrence risks in future pregnancies.
If a proband is affected, the parents must be advised to also protect infant and young siblings of the patient until a laboratory diagnosis can be made of their status.
Patients must be advised to refrain from exposure to environmental mutagens such as tobacco smoke.
Patients should be advised about the availability of DNA banking. This allows for future testing with yet undeveloped technology, especially for patients for whom clinical molecular genetic testing is not yet available or has not confirmed the diagnosis.
Differentials
Differential diagnoses for all chromosomal breakage syndromes are as follows:
-
Rothmund-Thomson syndrome
-
Erythropoietic protoporphyria
-
Toni-Fanconi syndrome not associated with cystinosis
-
Dyskeratosis congenita
-
Congenital porphyria
-
Hartnup disease
Differential diagnoses specifically for Fanconi anemia are as follows:
-
Ataxia telangiectasia
-
Bloom syndrome
-
Kindler syndrome
-
Thrombocytopenia with absent radii
-
VACTERL syndrome
-
Nijmegen breakage syndrome
Differential diagnoses specifically for ataxia telangiectasia are as follows:
-
Bloom syndrome
-
Generalized essential telangiectasia
-
Hereditary hemorrhagic telangiectasia
Differential diagnoses specifically for xeroderma pigmentosum are as follows:
-
Cockayne syndrome
-
Trichothiodystrophy
-
Cerebral-ocular-facial-skeletal syndrome
-
Carney complex
-
Kindler syndrome
Organizations And Support Groups
See the list below:
-
AT Children's Project, 668 S Military Trail, Deerfield Beach FL 33442-3023; (800) 5-HELP-AT, (954) 481-6611; info@atcp.org
-
A-T Medical Research Foundation, 5241 Round Meadow Rd, Hidden Hills CA 91302
-
The A-T Project, 3002 Enfield Rd, Austin TX 78703-3605; (512) 472-4892; A-Tproject@austin.rr.com
-
Genetic Alliance, Inc, 4301 Connecticut Ave NW, Suite 404, Washington DC 20008-2369; (202) 966-5557; info@geneticalliance.org
-
National Ataxia Foundation, 2600 Fernbrook Ln, Suite 119, Minneapolis MN 55447; (763) 553-0020; naf@ataxia.org
-
Bloom Syndrome Registry, Department of Pediatrics and Microbiology, Cornell University Medical College, 1300 York Ave, New York NY 10021; (212) 746-3956; (516) 678-5000 x6217. Email: jlg2003@mail.med.cornell.edu; msanz@molloy.edu
-
The Milo Gladstein Foundation for Bloom's Syndrome, 7095 Hollywood Blvd #583, Los Angeles, CA 90028, email: info@milogladsteinfoundation.org; www.milogladsteinfoundation.org
-
Xeroderma Pigmentosum, Genetics Education Center, University of Kansas Medical Center
-
National Organization for Rare Diseases, National Organization for Rare Disorders, 55 Kenosia Avenue, PO Box 1968, Danbury CT 06813-1968; (800) 999-6673 (voicemail only), (203) 744-0100, (203) 797-9590; orphan@rarediseases.org
-
Xeroderma Pigmentosum Registry, University of Medicine and Dentistry of New Jersey, Department of Pathology, Medical Science Building, Room C-520, 185 South Orange Ave, Newark NJ 07103-2714; (973) 972-4405
-
Xeroderma Pigmentosum Society, Inc., 437 Syndertown Rd, Craryville, NY 12521; (877) XPS-CURE (877-977-2873) or (518) 851-2612; xps@xps.org
-
XP Family Support Group, 8375 Folsom Blvd Suite 201, Sacramento CA 95826; (949) 218-9401; in United Kingdom
-
XP Skin Cancer Foundation, 5060 Taylor Rd #1, Cleveland OH 44128; (888) 254-0041
-
Fanconi Anemia Research Fund, Inc., 1801 Willamette St, Suite 200, Eugene OR 97401; (800) 828-4891, (541) 687-4658; info@fanconi.org
-
Fanconi Anemia Cell Repository, Department of Medical and Molecular Genetics, Oregon Health & Science University, 3181 SW Sam Jackson Park Rd L103, Portland OR 97201; (503) 494-6888
-
Face of a boy with ataxia-telangiectasia. Apparent ocular telangiectasia.
-
A young patient with Bloom syndrome showing the typical photodistributed erythema on the face. Courtesy of James L. German III, MD.
-
A 3-year-old patient with Fanconi anemia. Note the multiple birth defects, including short stature, microcephaly, microphthalmia, epicanthal folds, dangling thumbs, site of ureteral reimplantation, congenital dislocated hips, and rocker bottom feet. (Alter BP, Young NS. The bone marrow failure syndromes. In: Nathan DG, Oski FA, eds. Hematology of Infancy and Childhood, 4th ed. Philadelphia, PA: WB Saunders, Inc, 1993: 216-316.)
-
Face of a toddler with xeroderma pigmentosum, representative of an early stage of the disease. Note the freckling and the scaling. Courtesy of Neil S. Prose, MD, Duke University Medical Center, Durham, North Carolina.
Tables
Syndrome |
Gene Symbol: Chromosome Band |
Hypersensitivity: Immunodeficiencies |
Chromosome Breakage |
Cancer Risk |
Ataxia telangiectasia |
ATM: 11q22.3 (11q22–11q23 Human Genome Organization) |
Roentgentherapy, ionizing radiation, radiomimetic compounds: decreased immunoglobulin (Ig)A, IgG2, IgG, IgE |
TCRA: 14q11 TCRB: 7q35 IGH: 14q32 No increased sister chromatid exchanges (SCE), but increased gaps and breaks, nonhomologous interchanges |
The risk of neoplasia is 38% (85% are leukemias and lymphomas [B-cell]). Young children have acute lymphoblastic leukemia of T-cell origin. Older children have T-cell leukemia. The risk for other cancers is increased 4-fold (2-fold to 3-fold increased risk for breast cancer in carriers). |
Bloom syndrome < 1% of mutations |
BLM: 15q26.1 |
UV, cancer chemotherapeutic agents: Decreased IgA and IgM, and/or IgG |
SCE 12-15 times higher, homologous interchanges, increased gaps and breaks |
The risk for leukemia, lymphoma, adenocarcinoma, and other cancers is increased 5-fold to 8-fold. |
Fanconi anemia |
FANCA: 16q24.3 FANCB: Xp22.31 FANCC: 9q22.3 FANCD1/BRCA2: 13q12.3 FANCD2: 3p25.3 FANCE: 6p22-p21 FANCF: 11p15 FANCG/XRCC9: 9p13 FANCH = FANCA FANCI: 15q25-q26 FANCJ/BRIP1/BACH1: 17q23 FANCL/FAAP43: 2p16.1 FANCM/KIAA1596: 14q21.3 FANCN/PALB2: 16p12 FANCO/RAD51C: 17q22 FANCP/SLX4: 16p13.3 |
UV, chemotherapeutic agents for immunosuppression, diepoxybutane, mitomycin C, other DNA-crosslinking agents |
Increased gaps and breaks, 30% between nonhomologous chromosomes, clones with translocations |
For aplastic anemia, the risk is increased for pancytopenia, leukemia, acute myeloid leukemia, squamous cell carcinoma, medulloblastoma, Wilms tumor, breast cancer, and liver cancer. |
Xeroderma pigmentosum |
XPA: 9q22.3 XPB/ERCC3: 2q21 XPC: 3p25 XPD/ERCC2: 19q13.2-q13.3 XPE/DDB2: 11p12-p11 XPF/ERCC4: 16p13.3-p13.13 XPG/ERCC5: 13q33 XPV/POLH: 6p21.1-p12 ERCC1: 19q13.2-q13.3 |
UV XPG group may be sensitive to X-rays |
Increased gaps and breaks, increased SCE after UV |
The risk is increased 1000-fold for squamous cell carcinoma, basal cell carcinoma, malignant melanoma, and fibrosarcoma and is increased 10-fold to 20-fold for other tumors. |
Populations Affected |
ATM Mutation |
Effect or Comments |
|
Worldwide Amish, Turkish, Italian, German, Brazilian, Polish |
1563delAG |
Most common |
|
African American, and Korean |
L546V |
Increased risk of breast cancer |
|
American |
7271T>G5762ins137nt 8494C>T 5762ins137nt |
Kinase-dead mutation, milder phenotype Milder phenotype Slower neurologic deterioration |
|
Chinese |
G449A G204X R2227C |
Homozygous |
|
English/Irish, Polish |
7010delGT |
... |
|
German, Polish, American-Hispanic |
IVS20–579delAAGT |
... |
|
Iranian and Polish |
381delA |
Truncated protein in 23 of 25 carriers |
|
Italian, Polish |
8545C>T |
... |
|
Japanese, Polish |
742C>T |
... |
|
Korean |
IVS21+1049T>C IVS34+60G>A 3393T>G |
Increased risk of breast cancer, especially in premenopausal women; a genotypic polymorphism may play a role in breast cancer |
|
Mennonite, Polish, Danish, Norwegian, American Hispanic, German, Russian |
5932G>T |
... |
|
Philippine, Turkish, Polish |
5712insA |
... |
|
Polish, Danish, American Hispanic, Brazilian, Portuguese |
IVS53–2A>C |
Most common Polish mutation |
|
Polish, Swedish, German, French |
6095G>A |
Most common Polish mutation |
|
Spanish, Polish |
5188C>T |
... |
|
Childhood T-cell acute lymphoblastic leukemia |
S707P; F858L; P1054R; L1472W; Y1475C |
Found more often in T-cell acute lymphoblastic leukemia, with higher WBC count and increased risk of relapse |
|
Gene |
Locus |
Population |
Mutation |
Comments |
FANCA, 60-70% of mutations |
16q24.3 |
Northern European Israeli Arab |
3788-3790del 1115-1118del exons6-31del IVS42-2A>C |
Nuclear core complex protein |
FANCB, 2% of mutations |
Xp22.31 |
... |
1838insT 10693del3314 1650delT 811insT |
Nuclear core complex protein, X-linked inheritance, X-inactivation skewed towards mutant allele in female carriers, some cases present as VACTERL-H |
FANCC, 14% of mutations |
9q22.3 |
Ashkenazim Japanese Northern European Northern European Southern Italian |
IVS4+4A>T R548X, L554P, R185X, 322delG, Q13X |
Nuclear core complex protein, milder phenotype in Japanese, birth defects and earlier hematologic abnormality, congenital abnormality and later bone marrow failure |
FANCD1/BRCA2 – ~3% of mutations |
13q12.3 |
Ashkenazim Ashkenazi/ Lithuanian Latin American African American |
7691insAT 9900insA 6174delT/ C3069X 6174delT/ 886delGT I2490T/ 5301insA Q3066X/ E1308X |
Early-onset leukemia and solid tumors (breast cancer, brain tumors, medulloblastoma) |
FANCD2, 3% of mutations |
3p25.3 |
... |
... |
Frequent malformations, earlier and more severe hematologic manifestations |
FANCE, 3% of mutations |
6p22-p21 |
... |
... |
Nuclear core complex protein |
FANCF, 2% of mutations |
11p15 |
... |
... |
Nuclear core complex protein |
FANCG/XRCC9, 10% of mutations |
9p13 |
Korean/Japanese Brazilian French Canadian Northern European Israeli Arab |
IVS3+1G>C IVS8-2A>G IVS11+1G>C 1184-1194del 1794-1803del IVS4+3A>G |
Nuclear core complex protein |
FANCH=FANCA |
... |
... |
... |
... |
FANCI, 1% of mutations |
15q25-q26 |
... |
... |
Undergoes ubiquination and interacts with FANCD2 |
FANCJ/BRIP1/BACH1, 2% of mutations |
17q23 |
Many Argentinian |
R798X Y800X W647C, R707C |
Helicase that interacts with FANCD2 and BRCA1 |
FANCL/FAAP43,< 1% of mutations |
2p16.1 |
... |
... |
Ubiquitin protein ligase, nuclear core complex protein |
FANCM/KIAA1596,< 1% of mutations |
14q21.3 |
... |
... |
ATP-dependent RNA helicase, nuclear core complex protein |
FANCN/PALB2< 1% of mutations |
16p12 |
... |
... |
Early-onset leukemia and solid tumors, partner and localizer of BRCA2 |
FANCO/RAD51C< 1% of mutations |
17q22 |
... |
... |
DNA repair protein |
FANCP/SLX4< 1% of mutations |
16p13.3 |
... |
... |
Structure-specific endonuclease subunit |