Practice Essentials
Otto Werner originally defined Werner syndrome (WS) in 1904 on the basis of sclerodermalike, thin, tight skin and bilateral cataracts. WS is also known as progeria adultorum, progeria of the adult, and pangeria. WS is the most common of the premature aging disorders. WS and several other progeroid syndromes are epigenetically distinct disorders. [1]
Progeria can also refer to Hutchinson-Gilford syndrome, which is described as a lamin A gene defect and has onset early in life. The term progeria is derived from a Greek term meaning "before old age." See the images below. This syndrome represents an important model for aging, possibly allowing improved understanding of mechanisms and therapeutics of human aging. [2]
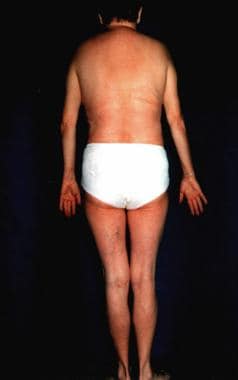
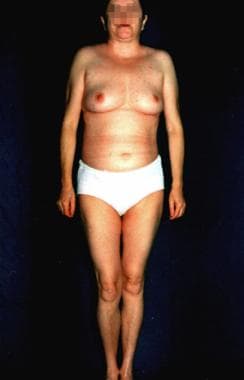
The aging process involves increasing errors in the mitotic machinery of dividing cells in the postreproductive stage of life; therefore, WS serves as a model for studying the aging process in vivo and in vitro. [3] However, many organs in patients with WS prematurely undergo changes that are usually associated with aging. No intellectual disability is observed.
WS is a premature aging disorder that may serve as a model of normal human aging. [4] In vivo aging is linked with accelerated aging of fibroblasts in culture, possibly due to the genomic instability, a hallmark of WS. Genome instability activates stress kinases, implying that kinase inhibitors may form the basis of antiaging therapies for individuals with WS.
Prognosis
The prognosis is unfavorable. The mean survival for patients with Werner syndrome (WS) is 46 years. Death usually occurs when patients are aged 30-50 years because of atherosclerosis or malignant tumors. Adroit medical management may enhance life expectancy; one patient was described who survived until dying of acute heart failure at age 68 years. [5]
Presentation
In young adults, mutation in the Werner syndrome (WS) gene is believed to be associated with clinical symptoms typically found in elderly individuals.
The most important feature of WS is healthy development in the patient's first decade of life.
Adult progeria is usually diagnosed on the basis of characteristic clinical features and typical concomitant diseases.
The hallmark of this syndrome is a striking disproportion between the patient's real age and the patient's appearance.
In general, this is an adult-onset disorder, with the earliest sign the lack of a growth spurt during adolescence. [6] A prematurely aged appearance with gray hair and sclerodermatous cutaneous changes begins in the 20-30s in association with cataracts, diabetes mellitus, atherosclerosis, cancers, and osteoporosis.
Metabolic abnormalities, including insulin resistance, may not be a pivotal part at disease onset. [7]
Also see Physical Examination and Complications.
Diagnostics
Both genetic testing and clinical criteria may not establish the diagnosis is some people. Gene testing can be salient to confirm autosomal recessive inheritance. [6] Also see Laboratory Studies.
Histologic findings
The skin and subcutaneous tissue of the extremities, especially where it is taut on clinical examination, shows epidermal thinning, loss of rete ridges, and dermal fibrosis with or without collagen hyalinization. Pilosebaceous structures are not well formed. Newly synthesized hyalinized collagen tends to replace the subcutaneous fat. No inflammatory infiltrate is usually evident.
Treatment
Specific treatment for Werner syndrome (WS) does not exist. Radiotherapy is contraindicated in this radiosensitivity syndrome. [8] Only symptomatic treatment of related disturbances is recommended. Results of a 2010 study on mice suggest that vitamin C supplementation could be beneficial for patients with WS. [9] Preclinical results suggest that everolimus may eventually prove to be of clinical benefit. [10]
Pathophysiology
Werner syndrome (WS) is an autosomal recessive disorder that affects connective tissue throughout the body. This segmental progeroid syndrome is caused by null mutations at the WRN locus, [11] which codes for a member of the RecQ family of DNA helicases. Enzymes in this group unwind double helix RNA and DNA. The protein is likely to be involved in the response to DNA damage during replication, as well as in the replication and transcription processes. The disease is connected with excessive synthesis of collagen types I and III, which is dependent on elevated messenger RNA (mRNA) levels. A Japanese WS patient was delineated with compound heterozygous WRN mutations, including a novel heterogeneous c.1720+1G>A substitution plus the most frequent heterogeneous c.3139-1G>C substitution among Japanese, yet with no malignancies detected despite the patient being aged 43 years. [12]
Targeted long-read sequencing may identify missing pathogenic variants in unsolved Werner syndrome cases. [13]
Skin fibroblasts in WS patients demonstrate characteristics of cells in conditions of stress with slow growth rates, an elongated cell cycle, and an altered morphology that suggests stress-induced premature senescence transduced in part by the p38α MAP kinase signaling pathway. This p38 involvement has been documented in WS cells. [14]
More than 70 disease-causing mutations have been described, the majority being stop codon mutations, splice mutations, or small ins/del-producing truncations of the protein and/or non-sense–mediated decay of mutant mRNA. [6] Two novel WRN mutations were described in patients of South Asian ancestry. [15] Another patient had a homozygous novel 1bp-deletion in exon 19 of WRN, 2426/27delG, causing frameshift and protein truncation R809SfsX2. [16]
The collagenase level is also increased several times. However, a child may have characteristics of WS but with no identifiable mutation in the WRN gene. Such cases are classified as atypical WS. [17] A paper from 2010 reported 18 new mutations, including 2 genomic rearrangements, a deep intronic mutation resulting in a novel exon, a splice consensus mutation leading to utilization of the nearby splice site, and 2 rare missense mutations. [18]
WS has been classified as a member of the RecQ family of DNA helicases implicated in the resolution of DNA structures leading to the stall of replication forks. [19] Fragile sites may be DNA regions particularly sensitive to replicative stress. Evidence was recently presented of a crucial role for a helicase in protecting cells against chromosome breakage at normally occurring replication fork-stalling sites.
Human progeroid syndromes are linked with mutations in single genes accelerating some, but not all, features of normal aging. [20] Most are associated with defects in genome maintenance.
LMNA (lamin A/C) gene mutation has been described with atypical Werner syndrome, with the severe metabolic complications, the extent of the lipodystrophy being linked with A133L mutation in the LMNA gene. [21, 22]
Epidemiology
Frequency
United States
Werner syndrome (WS) is a rare disorder. The estimated incidence is 1 case in 1 million individuals.
International
WS is more common in Japan and Sardinia than in other regions. [23] About 1000 cases are reported in the world; more than 800 of these cases are in Japan. Approximately 30% of 116 Japanese patients were noted to be the product of a consanguineous marriage. [24]
Race-, sex-, and age-related information
No racial predilection is reported. Both sexes are affected equally.
The onset of WS might occur in individuals in their mid teens, or it may be delayed until an individual is as old as 30 years. The average age of patients at the time of diagnosis is 37 years.
-
Werner syndrome.
-
Werner syndrome.





