Practice Essentials
Short stature may be the normal expression of genetic potential, in which case the growth rate is normal, or it may be the result of a condition that causes growth failure with a lower-than-normal growth rate. [1] Growth failure is the term that describes a growth rate below the appropriate growth velocity for age (see image below).
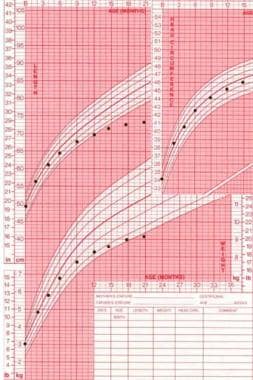 Growth failure in length and weight with a normal head circumference in an infant with growth hormone deficiency.
Growth failure in length and weight with a normal head circumference in an infant with growth hormone deficiency.
A child is considered short if he or she has a height that is below the fifth percentile; alternatively, some define short stature as height less than 2 standard deviations below the mean, which is near the third percentile. Thus, 3-5% of all children are considered short. Many of these children actually have normal growth velocity. These short children include those with familial short stature or constitutional delay in growth and maturation, which are normal nonpathologic variants of growth. In order to maintain the same height percentile on the growth chart, growth velocity must be at least at the 25th percentile. When considering all children with short stature, only a few actually have a specific treatable diagnosis. Most of these are children with a slow growth velocity.
Pathophysiology
The most rapid phase of human growth is intrauterine. Following birth, a gradual decline in growth rate occurs over the first several years of life. The average length of an infant at birth is about 20 inches, the length at age 1 year is approximately 30 inches, the length at age 2 years is approximately 35 inches, and the length at age 3 years is approximately 38 inches. After age 3 years, linear growth proceeds at the relatively constant rate of 2 inches per year (5 cm/y) until puberty.
Normal growth is the result of the proper interaction of genetic, nutritional, metabolic, and endocrine factors. To a large extent, growth potential is determined by polygenic inheritance, which is reflected in the heights of parents and relatives. Secretion of growth hormone (GH) by the pituitary is stimulated by growth hormone–releasing hormone (GHRH) from the hypothalamus. GHRH also stimulates somatotroph proliferation. Another signal, which is stimulated by certain growth hormone–releasing peptides (GHRPs), may be present; the receptor for the GHRPs has been identified, and ghrelin, the natural ligand for these receptors, has been identified. The GHRH receptor is a cell surface-associated seven membrane-spanning domain protein linked to a G protein (Gs). It stimulates intracellular cAMP production after ligand-induced activation.
Ghrelin (from the word ghre, a root word in proto-Indo-European languages meaning grow), is unique in that it is a small polypeptide modified at the third amino acid (serine) by esterification of n-octanoic acid. Ghrelin is a gastrointestinal peptide (synthesized in the stomach) which specifically induces GH secretion. The ghrelin receptor is expressed on the anterior pituitary. Somatostatin secreted by the hypothalamus inhibits growth hormone secretion.
When growth hormone pulses are secreted into the systemic circulation, insulin-like growth factor (IGF)–1 is released, either locally or at the site of the growing bone. Growth hormone circulates bound to a specific binding protein (GHBP), which is the extracellular portion of the growth hormone receptor. IGF-1 circulates bound to one of several binding proteins (IGFBPs). The IGFBP that most depends on growth hormone is IGFBP-3.
Etiology
The following are possible causes of growth failure (slow growth velocity):
-
Familial short stature: Children with familial short stature have a history of parents with short stature. They have a normal growth velocity (thus, they do not exhibit true growth failure). Bone age is not delayed. These children have puberty at a normal time and most often finish their growth with a short adult height. [2]
-
Constitutional delay in growth and maturation: This entity is sometimes called delayed puberty. Children with constitutional delay have a normal birth weight, and during the first year of life, their growth slows. For most of the period of linear growth (approximately age 3 y to puberty), they maintain an adequate growth velocity. Bone age is usually delayed, and puberty is late, giving a longer time for prepubertal growth, which usually results in a normal adult height. Children with constitutional delay may have a family history of the same. Usually, these children do not exhibit growth failure (a slow growth velocity); however, a period of slow growth velocity usually occurs during the first year of life, and, just before the onset of puberty, growth velocity is again slow (especially when compared with peers who are in the midst of their pubertal growth spurt). [3]
-
Malnutrition: Worldwide, malnutrition is probably the most common cause of growth failure and is usually poverty related. In developed countries, nutritional deficiencies are more often the result of self-restricted nutrient intake. Often, poor weight gain is more striking than short stature.
-
Chronic disease, systemic disorders
Nervous system: Microcephaly may be a feature.
Circulatory system: Cyanotic heart disease may be present.
Gastrointestinal system: Gluten enteropathy (celiac disease), ulcerative colitis, or regional enteritis (Crohn disease), disorders involving the liver may be present. In inflammatory bowel disease (in particular, Crohn disease), the growth failure may be apparent before other symptoms appear.
Renal disease: Chronic renal failure, renal tubular acidosis. In children, growth failure may precede the diagnosis of chronic renal failure.
Lungs: Cystic fibrosis or severe asthma may be present.
Connective tissue/rheumatologic problems: Conditions such as dermatomyositis or systemic-onset juvenile idiopathic arthritis (JIA) may be present.
-
Psychosocial dwarfism
-
Chromosomal abnormalities: In particular, Turner syndrome (45,X) and Down syndrome (trisomy 21) have growth failure as a part of the syndromes. Growth charts specific for these syndromes are available. Short stature homeobox-containing gene (SHOX) mutations, haploinsufficiency, or complete absence are associated with growth retardation (OMIM #300582). The SHOX gene is found in the pseudoautosomal region of the X and Y chromosomes. Individuals with SHOX mutation tend to have mesomelic growth retardation (shorter forearms and lower legs), Madelung deformity of the forearm (focal dysplasia of the distal radial physis), cubitus valgus, high arched palate and muscular hypertrophy (short, stocky appearance). SHOX mutations are present in approximately 1-4% of patients who would otherwise have been classified under the category of idiopathic short stature [4] .
-
Other syndromes (nonchromosomal): Syndromes that have growth failure as a feature include Noonan syndrome, Russell-Silver syndrome, and Prader-Willi syndrome.
-
Target tissue defects
Intrauterine growth retardation: The category of intrauterine growth retardation describes children who have birth weights less than 5.5 lb at full term or who are small for gestational age (SGA) if born preterm. Numerous etiologies for this condition are contained in this category, including fetal alcohol syndrome and placental insufficiency syndromes. In some of these conditions, spontaneous "catch-up" growth occurs, while in others, growth rate remains slow. Overall, 10% of children who are SGA have not caught up in growth by age 2 years.
Bone and cartilage disorders: The most common disorder of bone and cartilage is achondroplasia, which is recognizable by frontal bossing, lumbar lordosis, and short limbs. Other skeletal disorders are less easily recognized, such as hypochondroplasia, which may be diagnosed radiologically. Patients with hypochondroplasia also have short limbs, but the disproportion is subtle and may be apparent only with careful measurements of arm span and US and LS. Both of these disorders are due to mutations of the fibroblast growth factor receptor 3.
-
Endocrine causes
Thyroid hormone deficiency (hypothyroidism): Thyroid hormone is absolutely necessary for normal growth. With hypothyroidism, the growth rate is extremely slow, and with replacement of thyroid hormone, catch-up growth is rapid. Although hypothyroidism is often suspected based on history and physical examination findings, cases have also been reported in which the signs and symptoms are subtle. Because of the possibility of subtle signs, evaluation of thyroid hormone levels in all children with slow growth is advised.
Growth hormone deficiency: Children who are growth hormone deficient have normal proportions but may appear younger than their age. They have delayed skeletal maturation. Although growth hormone deficiency may be suspected because of damage or malformation of the pituitary gland, in most children diagnosed with growth hormone deficiency, the etiology is idiopathic.
Growth hormone insensitivity (primary IGF-1 deficiency): Sometimes called Laron dwarfism, this disorder appears to be similar to growth hormone deficiency, except that large amounts of growth hormone are produced but levels of IGF-1 are low. This is a rare condition, except in populations where the gene is present with a greater frequency (eg, in Ecuador).
Glucocorticoid excess (Cushing syndrome, Cushing disease): Children with glucocorticoid excess almost always have growth failure as part of the presentation.
Androgen excess: When prepubertal children are exposed to excessive amounts of androgen, the growth velocity increases in the short term, but epiphyseal fusion occurs early, resulting in premature slowing of growth velocity, usually resulting in a short adult height. Causes of androgen excess include exposure to exogenous androgen, precocious puberty, and congenital adrenal hyperplasia.
Epidemiology
United States statistics
In 1994, Lindsay et al studied 114,881 school children in Utah. [5] After 1 year, 79,495 of the original group were available for evaluation. Of these, 555 (0.7%) had heights that were below the third percentile and a growth rate that was less than 5 cm/y. When examined further, causes for short stature within this group of children included familial short stature (37%), constitutional delay (27%), a combination of familial short stature and constitutional delay (17%), other medical causes (10%), idiopathic short stature (5%), growth hormone deficiency (3%), Turner syndrome (3% of girls), and hypothyroidism (0.5%).
International statistics
Several studies have been conducted to determine the frequency of various causes of short stature. In 1974, Lacey and Parkin evaluated children in Newcastle upon Tyne in England. [6] They studied 2256 children, 111 of whom were below the third percentile in stature. Of the 98 children that they were able to examine, only 16 had evidence of organic disease causing their short stature. Diagnoses included Down syndrome, cystic fibrosis, chronic renal insufficiency, growth hormone deficiency, juvenile rheumatoid arthritis (treated with glucocorticoid), and Hurler syndrome.
Across low- and middle-income countries, the estimated prevalence of stunting (the most widespread indicator of child growth failure) decreased from 36.9% in 2000 to 26.6% in 2017. By 2017, the areas with the highest prevalence of stunting were found throughout much of the sub-Saharan Africa, Central and South Asia, and Oceania regions. [7]
Race- and sex-related demographics
There is no known racial predilection for growth failure; however, in large databases following children treated with growth hormone, [8] White children appear to be over-represented, compared with children of Asian or African descent. This observation is thought to be probably due to referral bias.
The sex distribution of children treated with growth hormone is about 3 boys for every girl. Recent work in this area suggests that this is mostly due to a referral bias, either from parents themselves or from the referring physician.
Prognosis
Prognosis for adult stature depends on the cause of the growth failure. Initiating therapeutic intervention is important before the patient has closure of the epiphyses with the concomitant finishing of the growth process. If a diagnosis of hypothyroidism or growth hormone (GH) deficiency is made, replacement of the deficient hormone usually results in a period of rapid catch-up growth, with subsequent normal growth until epiphyseal fusion.
Morbidity/mortality
Short stature has been thought to have far-reaching effects on psychological well-being, including poor academic achievement (despite normal intelligence, healthy family dynamics, and high socioeconomic status) and behavioral problems (eg, anxiety, attention-seeking actions, poor social skills).
Morbidity related to the underlying cause of the growth failure may also be observed. Some studies involving children who have not been seen in a clinic that treats short stature (and, therefore, may represent a different patient population) have challenged the notion that short stature has psychological implications. At the present time, this issue is not completely resolved.
Mortality rates in children with growth failure relate to the underlying cause of the growth failure. Mortality is not related to growth failure itself; rather, it is related only to the cause of the growth failure.
-
Growth failure in length and weight with a normal head circumference in an infant with growth hormone deficiency.






