Practice Essentials
Nephropathic cystinosis is an inherited (autosomal recessive) lysosomal storage disorder caused by defective transport of the amino acid cystine out of lysosomes. The stored cystine is poorly soluble and crystallizes within the lysosomes of many cell types, leading to widespread tissue and organ damage.
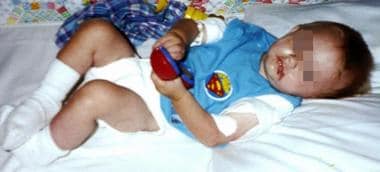
The image below depicts an infant at the time his cystinosis is diagnosed.
Signs and symptoms
Three types of cystinosis have been described based on the age at diagnosis and magnitude of cellular cystine deposition: infantile onset, adolescent onset, and adult onset. Patients with the infantile nephropathic form of cystinosis (the most common and the most severe) develop symptoms early in life and, if left untreated, develop end-stage kidney failure by late childhood.
Signs and symptoms of nephropathic infantile cystinosis include the following:
-
Multiorgan involvement: May be mild to severe
-
Polyuria, polydipsia, dehydration, vomiting, metabolic acidosis
-
Hypophosphatemic rickets
-
Constipation
-
Failure to thrive, poor/loss of appetite
-
Craves salty and hot and spicy foods; prefers specific food textures
-
May have recurrent bouts of fever and manifestations of heat intolerance
-
Left untreated, renal failure develops by age 7-10 years
Signs and symptoms of late-onset (intermediate) nephropathic cystinosis include the following:
More indolent disease than infantile form of the disease
-
Manifests most commonly in early adolescence; most diagnosed by age 12 years
-
Symptoms usually restricted to kidneys and eyes
-
Often does not develop complete Fanconi syndrome, but renal function deteriorates
-
End-stage renal disease within a few years of diagnosis, after age 15 years
The major complication of nephropathic cystinosis in patients older than 20 years is legal blindness, distal vacuolar myopathy, cerebral calcifications or atrophy, swallowing dysfunction, diabetes mellitus, and liver disease (eg, hepatomegaly, nodular degenerative hyperplasia).
Nonnephropathic cystinosis is considered a benign variant and is usually diagnosed by an ophthalmologist treating patients for photophobia, which may not begin until middle age and is not usually as debilitating as in the nephropathic form of the disease.
See Clinical Presentation for more detail.
Diagnosis
A typical cystinotic patient has pale blond hair and blue eyes, although the disease also occurs among dark-haired individuals with brown eyes. Examination in patients with nephropathic cystinosis (progressive disease) may also reveal the following features:
-
Children younger than 1 year: Usually growth retardation, rickets, metabolic acidosis, and other chemical evidence of renal tubular abnormalities (eg, increased renal excretion of glucose, amino acids, phosphate, and potassium)
-
As children age: Prominent failure to thrive; without specific therapy, children remain below the third percentile in both height and weight throughout life
-
By age 1-2 years: Corneal crystals
-
By age 3-4 years: Untreated, cornea packed with crystals, causing early photophobia
-
By age 7-10 years: Previously noted symptoms now more severe; increased proteinuria, progressive renal failure, increased photophobia, thyroid insufficiency
-
Adolescence (usually age 10-13 years): End-stage renal disease; good adherence to therapy slows down-progression of renal failure by several more years
-
Delayed sexual maturation; in males, hypogonadism and infertility
-
Second or third decade of life: Retinal damage
-
Third decade of life: Cerebral calcifications; muscular and swallowing difficulties
Laboratory tests
The following laboratory studies may be used to assess patients suspected of having cystinosis:
-
Serum electrolyte levels: To detect the presence of acidosis (hyperchloremic, normal anion gap) and severity of hypokalemia, hyponatremia, hypophosphatemia, and low bicarbonate concentration
-
Blood gases: To detect metabolic acidosis and the degree of respiratory compensation
-
Urine testing: Findings include low osmolality, glycosuria, and tubular proteinuria (including generalized amino aciduria)
-
Urine electrolyte levels: To detect the loss of bicarbonate and phosphaturia
-
Cystine levels in polymorphonuclear leukocytes or cultured fibroblasts (for fetuses: chorionic villi or cultured amniotic fluid cells): Confirms diagnosis of cystinosis
Other studies used in the evaluation of patients with suspected cystinosis include the following:
-
Slit-lamp examination of the eyes: Corneal and conjunctival crystals (pathognomonic)
-
Funduscopic examination: Possible presence of peripheral retinopathy that is more severe on the temporal side than on the nasal side
-
Biopsy of the kidney with histologic examination: Changes vary with disease stage
Imaging studies
-
Renal ultrasonography: Obtain in patients with elevated urine calcium excretion to rule out nephrocalcinosis
-
Radiography of kidneys, ureters, and bladder: To evaluate possible urinary tract calcifications in patients with hypercalciuria or as a diagnostic evaluation of severe abdominal pain
-
Computed tomography scanning and magnetic resonance imaging: To evaluate adult patients with infantile nephropathic cystinosis with central nervous system symptoms
See Workup for more detail.
Management
Cysteamine blunts the decline in renal function and improves the linear growth of children with nephropathic cystinosis, despite the fact that it does not ameliorate the defect in renal tubule transport. Oral therapy should be initiated as soon as the diagnosis is made.
Pharmacotherapy
Cysteine-depleting agents are used in the management of nephropathic cystinosis. Oral cysteamine therapy postpones the need for renal transplantation, whereas ophthalmic cysteamine is used to manage ocular complications such as corneal cystine crystals.
Indomethacin may limit water losses in patients with nephropathic cystinosis by both reducing the glomerular filtration rate and by sensitizing the collecting duct to the effects of antidiuretic hormone. It has been used to treat patients with cystinosis more commonly in Europe than in the United States. Careful monitoring of kidney function is required for this agent, because the glomerular filtration rate may be worsened by its administration. The ulcerogenic potential of indomethacin is its major drawback for treating cystinosis.
Treatment with recombinant human growth hormone improves growth velocity in young children with nephropathic cystinosis prior to renal replacement therapy. Growth hormone treatment is less effective for peripubertal/adolescent patients on renal replacement therapy.
Thyroid replacement is indicated in patients diagnosed with hypothyroidism.
Supportive therapy
Supportive care in the management of nephropathic cystinosis includes the following:
-
Replacement of urinary losses (eg, hydration, potassium and bicarbonate supplementation, vitamin D and phosphate supplementation; carnitine)
-
Management of volume depletion/dehydration states
Surgery
Kidney transplantation in patients with infantile cystinosis corrects kidney failure and prolongs survival (the donor parenchymal cells are not homozygous for the genetic defect and are therefore able to transport cystine from the lysosomes). However, transplantation does not prevent progression of the disease in other nonrenal organs, and therapy with oral cysteamine is indicated in patients after kidney transplantation. Management of cystinosis with oral cysteamine must be initiated as soon as diagnosis of cystinosis is made.
Some patients with severe gastroesophageal reflux and cystinosis may require gastric/jejunal tube placement or Nissen fundoplication to achieve optimal nutrition.
See Treatment and Medication for more detail.
Background
Nephropathic cystinosis is an inherited (autosomal recessive) lysosomal storage disorder caused by defective transport of the amino acid cystine out of lysosomes. The stored cystine is poorly soluble and crystallizes within the lysosomes of many cell types, leading to widespread tissue and organ damage.
Three types of cystinosis have been described based on the age at diagnosis and magnitude of cellular cystine deposition: infantile onset, adolescent onset, and adult onset. Patients with the infantile nephropathic form of cystinosis (the most common and the most severe) develop symptoms early in life and develop end-stage kidney failure by late childhood, if untreated.
Specific therapy with a drug that allows for removal of cystine from its lysosomal accumulation has been associated with marked improvement in the outlook for kidney function and quality of life in patients with nephropathic cystinosis.
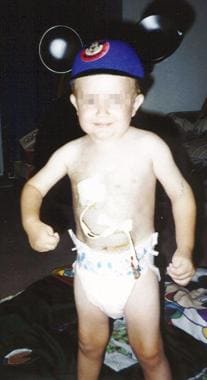 The same child as in the previous images, at age 4 years, on total parenteral nutrition via central line.
The same child as in the previous images, at age 4 years, on total parenteral nutrition via central line.
 The same child as in the previous images, at age 9 years, off total parenteral nutrition for 1 year and tolerating oral intake.
The same child as in the previous images, at age 9 years, off total parenteral nutrition for 1 year and tolerating oral intake.
Pathophysiology
Ingested protein enters the lysosome, where acid hydrolases degrade it to its component amino acids, including cysteine. Within the lysosome, cysteine is readily oxidized to cystine (a disulfide of the amino acid cysteine). In healthy individuals, both cystine and cysteine can normally enter the cytoplasm, where cystine is rapidly converted to cysteine by the reducing agent glutathione. Cytoplasmic cysteine is incorporated into protein or degraded to inorganic sulfate for excretion.
Cystinosis is caused by mutations in the gene that encodes cystinosin, the cystine-lysomal exporter. Because of the defect in cystinosin, cystine cannot leave the lysosomes and is accumulated there as birefringent, hexagonal, or rectangular crystals within cells of various organ systems.
In the infantile nephropathic form of cystinosis, the kidney is affected early in life by cystine crystals deposited in proximal tubule cells, presenting as Fanconi syndrome, usually between age 6-12 months. Proximal tubules are particularly susceptible to the adverse effects of cystine accumulation. Fanconi syndrome is characterized by wasting of substances not reabsorbed in the proximal tubule, including sodium, potassium, phosphate, calcium, magnesium, bicarbonate, and others. Metabolic acidosis and electrolyte disturbances ensue and contribute to the stunting of growth in children with cystinosis. Cystinosis is the most common inherited cause of Fanconi syndrome.
Etiology
All forms of cystinosis have autosomal recessive patterns of inheritance. Cystinosis is caused by a defect in transport of cystine across the lysosomal membrane due to defective function of the lysosomal membrane protein cystinosin, resulting from mutations of the cystinosis gene (CTNS). CTNS resides on chromosome 17p13. The CTNS gene has 12 exons, the last 10 of which code for cystinosin.
Cystinosin (an integral lysosomal membrane protein) has 367 amino acids and 7 transmembrane domains. In nephropathic cystinosis patients, CTNS mutations can cause either an absence of cystinosin or a disruption of transmembrane domains and loss of protein function, leading to inhibition of cystine transport through the lysosomal membrane (which is carrier-dependent). More than 80 different CTNS mutations (missense, nonsense, splice-site, deletion, and promoter mutations) are described in patients with nephropathic cystinosis; the most common are 57 kilobases (kb) (approximately 60% of the mutations in US patients).
Mutations of CTNS that affect functionally unimportant regions of cystinosin account for a milder clinical course. The various CTNS mutations can explain why patients have a wide spectrum of clinical symptomatology.
The parents of patients with cystinosis are obligate heterozygotes for cystinosis; they each carry a single gene for the disease. Individuals heterozygous for cystinosis have never been reported to have cystine crystals in any tissue or cell. Despite the clinically normal appearance of individuals who are heterozygous for cystinosis, their polymorphonuclear cells contain an increased amount of cystine.
Late-onset (intermediate) cystinosis appears to be due to the inheritance of a mutation known to cause infantile disease in one allele and a relatively less clinically severe mutation in the other or due to the inheritance of a relatively less severe mutation in both alleles.
Epidemiology
United States data
In North America, the incidence of infantile nephropathic cystinosis is 1 case per 100,000-200,000 live births [1] ; an estimated 400 individuals in the United States have cystinosis. Approximately 15 new cases of cystinosis are diagnosed each year in the United States.
International data
The incidence of cystinosis is higher in certain subpopulations. France's Brittany province has an estimated incidence of 1 case per 25,909 population; the incidence in the rest of France is 1 case per 326,440 population.
Race-, sex-, and age-related demographics
Cystinosis is often considered a disease of fair-skinned individuals of European descent; however, it is known to occur in Blacks, Hispanics, and other races around the globe.
The male-to-female ratio among cystinotic children has been reported to be 1.4:1.
Patients with infantile nephropathic cystinosis develop initial symptoms in infancy, frequently when younger than 1 year. In the late-onset (adolescent) form, symptoms are evident by age 8-12 years, and the progression is slower. The adult form of cystinosis does not include renal involvement and is limited to the eye (ocular cystinosis).
Prognosis
Morbidity/mortality
Medical perceptions regarding the complications and outcome of cystinosis have changed over the years. Prior to the availability of renal transplantation, infantile cystinosis was considered a fatal disease, with a lifespan of approximately 10 years. The natural history of the disease has changed dramatically since the introduction of renal transplantation and cysteamine. Patients with infantile cystinosis now survive into even the fifth decade of life.
The study results from a large European cohort showed improved survival of renal function and better patient and graft survival in children with nephropathic cystinosis who received renal replacement therapy compared with those who did not. The 5-year survival rate after the start of renal replacement therapy improved from 86.1% (prior to 1990) to 100% (since 2000). [2]
Cysteamine, introduced in the early 1980s, was shown to blunt the decline in renal function and improve linear growth in these children, despite the fact that it does not ameliorate the defect in renal tubule transport. However, the increased life expectancy afforded by the progress in medical and surgical treatment was accompanied by the development of serious complications due to the continuous accumulation of cystine in nonrenal organs, including the eye, thyroid, brain, liver, pancreas, and muscle. However, many patients survive into the third or fourth decade of life, some even survive into the fifth decade of life and are able to pursue fulfilling lifestyles.
Complications
Before renal transplantation, nonrenal complications of infantile cystinosis include photophobia, corneal crystals, hypothyroidism, and short stature.
After renal transplantation, nonrenal complications include photophobia, corneal crystals, hypothyroidism, polyneuropathy, distal myopathy, CNS abnormalities, renal stones, and diabetes mellitus. [3] Among the more unusual complications noted in the literature are pulmonary fibrosis, nodular degenerative hyperplasia of the liver, and hearing loss.
In a study of nonrenal complications in 21 patients with cystinosis (aged 6 months to 29 years), 14 patients had hepatomegaly, 11 had portal hypertension, and 6 had gastroesophageal reflux. A lack of adherence to treatment was associated with complications from cystine accumulation in nonrenal organs. [4]
Most male patients with cystinosis are infertile as a result of azoospermia due to obstructive causes. [5] In contrast, female patients with cystinosis seem to have normal fertility; however, they are at high risk for complications of pregnancy, including gestational diabetes, preeclampsia, and cephalopelvic disproportion. [6]
Patient Education
Because of the chronic and rare nature of the disease, allowing the parents of patients with cystinosis full access (eg, handouts to take home, library card) to current articles on the disease is critical. Parents may also join the Cystinosis Research Network, the Cystinosis Research Foundation, or the Cystinosis Foundation to become familiar with the disease, to exchange their knowledge, and to bond their children with peers who also have cystinosis.
-
An 8-month-old male infant at the time his cystinosis is diagnosed.
-
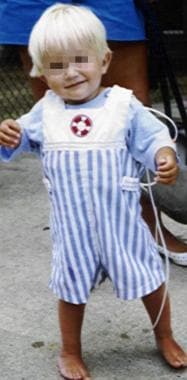
The same child as in the previous image, at age 20 months, fed via gastric tube.
-
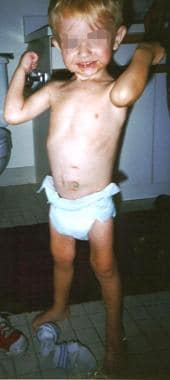
The same child as in the previous images, at age 3 years, fed via jejunal tube.
-
The same child as in the previous images, at age 4 years, on total parenteral nutrition via central line.
-
The same child as in the previous images, at age 9 years, off total parenteral nutrition for 1 year and tolerating oral intake.
Tables

What would you like to print?
- Optimal Management of Perianal Disease in Crohn's Disease: The Importance of a Multidisciplinary Approach
- Alzheimer’s Disease: The Cholinergic Revival
- Loneliness/Disease Link Debatable?
-
 5 Interesting Neurology Studies
5 Interesting Neurology Studies
-
Treating Deadly Disease in Utero Called 'Revolutionary' Advance
-
Positive Topline Results for Novel Parkinson's Treatment




