Background
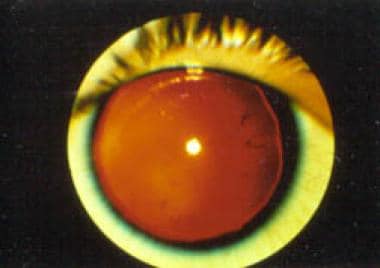
Patients with an unusual complex of congenital developmental abnormalities, such as aniridia (see the image below), genitourinary (GU) malformations, and intellectual disability (previously termed mental retardation), are at high risk (>30%) of having a Wilms tumor. At birth, the association is aniridia, GU malformations, and intellectual disability (AGR) syndrome. With the discovery of a Wilms tumor in these patients, the association is referred to as WAGR syndrome. These syndromes result from the loss of chromosomal material from the short arm of chromosome 11.
Aniridia, GU malformations, and intellectual disability are usually detected in the perinatal period, and patients with these conditions require careful long-term follow-up, both because of the consequences of the congenital defects and because of the potential development of a Wilms tumor. Early tumor detection has improved the long-term disease-free survival of children with WAGR syndrome.
Pathophysiology
WAGR syndrome affects the development of seemingly disparate areas of the body, including the kidney, the GU system, the iris of the eye, and the CNS. The pathogenic germline deletion of varying lengths of chromosomal material along the short arm of chromosome 11, including WT1 and PAX6, is the underlying defect. [1]
The constitutional loss of one allele of the Wilms tumor gene (WT1) results in GU anomalies and forms the first of 2 genetic events in the development of a Wilms tumor. [2] The product of the WT1 gene has zinc finger arrays that bind to specific DNA sequences, whereas the amino terminus appears to regulate transcription. Alterations to the remaining allele result in the development of a Wilms tumor, usually in early childhood. Meanwhile, the deletion of one copy of the PAX6 gene is responsible for aniridia. PAX6 plays a role in CNS development as well and may be responsible for the intellectual disability seen in a reported 75% of children with WAGR syndrome. [3]
The brain-derived neurotrophic factor (BDNF) gene is also located in the region of chromosomal loss associated with WAGR syndrome. [4] Loss of function of the BDNF gene in some patients with WAGR syndrome may produce obesity and hyperphagia. [5, 6] See the image below.
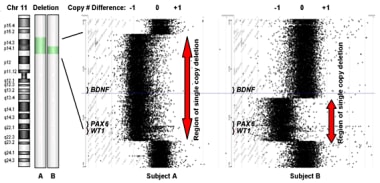 Subjects were categorized as BDNF haploinsufficient by comparative genomic hybridization. Subject A has a large deletion on chromosome 11 that removes one copy of the BDNF gene. Subject B has a smaller deletion that does not remove BDNF.
Subjects were categorized as BDNF haploinsufficient by comparative genomic hybridization. Subject A has a large deletion on chromosome 11 that removes one copy of the BDNF gene. Subject B has a smaller deletion that does not remove BDNF.
Etiology
WAGR syndrome is caused by the contiguous loss of chromosomal material from the short arm of chromosome 11.
The identification of the gene responsible for Wilms tumor did not occur until 1990, when 3 groups independently identified the WT1 gene on band 11p13. [7, 8, 9] Note the following:
-
The genetic structure includes 4 zinc-finger regions, which suggest that WT1 may be important in controlling the expression of other genes. Both the GU abnormalities and the development of a Wilms tumor in patients with WAGR syndrome are related to the loss of WT1 gene function. In adults, WT1 isoforms continue to be expressed in some kidney tissue.
-
A neighboring gene, PAX6, is responsible for the development of the iris. Deletion of the PAX6 gene as part of the band 11p13 deletion in patients with AGR or WAGR syndrome results in aniridia.
-
Deletion of the PAX6 gene, which plays a role in myelinization of the cerebral hemispheres during CNS development, may also be responsible for the intellectual disability seen in the WAGR association. An association between PAX6 abnormalities and diabetes may indicate that it plays a role in pancreatic development as well.
Patients with early bilateral Wilms Tumor suggests the possibility that they have a constitutional genetic defect that predisposes them to the development of a Wilms tumor, like that seen in WAGR syndrome. [10] The prezygotic loss of one of the WT1 alleles is followed by the loss of the second allele in infancy or early childhood (somatic).
Epidemiology
Race- and age-related demographics
The incidence of WAGR syndrome has not been determined. Wilms tumor occurs in approximately 8 per 1 million White children in the United States; the incidence is somewhat higher in Blacks.
Aniridia and/or GU abnormalities are usually detected while the baby is in the newborn nursery, and the diagnosis of AGR syndrome is considered at that time.
Wilms in WAGR
About 10-15% of patients with Wilms tumor have an underlying germline predisposition. [11, 12] In a US study of 3442 patients with Wilms tumor, only 26 (0.76%) presented with aniridia. [13] Wilms tumor occurs in more than 40-65% of patients with 11p13 deletions. [12]
Prognosis
Patients with WAGR syndrome have an excellent prognosis for long-term survival. Early detection seems to improve the outcome.
Life-limiting abnormalities include GU anomalies in the first year of life. Lifelong disabilities may include vision loss and intellectual disability.
In the approximately 30% of patients with AGR syndrome in whom a Wilms tumor develops, the prognosis is related to the histologic features and the stage of the tumor.
Wilms in WAGR
The overall survival rate of patients with Wilms tumor is excellent and is related to the histologic features of the tumor (favorable vs unfavorable) and the stage of the disease, as follows:
-
In stage I, the disease is localized to the kidney and is completely removed by surgery.
-
In stage II, the disease extends beyond the kidney and is completley removed by surgery.
-
In stage III, there is residual non-hematogenous disease confined to the abdomen.
-
In stage IV, distinct metastases are present.
-
In stage V, bilateral kidney involvement is present.
Across all patients with Wilms Tumor, overall survival is approximately 90%. However, certain patients fare less well including patients with stage IV anaplastic Wilms, with overall survival of < 50%. On AREN0534, patients with stage V tumors, some of whom had WAGR syndrome, had an overall survival rate of approximately 94.9%. [14]
GU abnormalities in WAGR
A wide variety of GU abnormalities are associated with WAGR syndrome; these include cryptorchidism, hypospadias, and renal and ureteral malformations. Streak ovaries and bicornuate uterus have been reported in females with AGR syndrome. The presence of pseudohermaphroditism should alert the clinician to the possibility of WT1 disorder (which includes the sydnrome previously known as Denys-Drash syndrome), a distinct diagnosis resulting from constitutional WT1 mutations.
Renal function in WAGR
Patients with WAGR have an increased risk of end stage renal disease. [15, 16]
Aniridia in WAGR
Aniridia results in decreased visual acuity, although the amount of vision loss varies. Aniridia has been associated with the development of glaucoma, probably due to the structural abnormalities of the anterior chamber of the eye. Cataracts have also been reported in these patients. Over time, scanning nystagmus develops in infants who are visually impaired. Other ocular abnormalities seen in these patients include corneal pannus and optic nerve hypoplasia.
Intellectual disability in WAGR
The cognitive function of patients with WAGR syndrome widely varies. The appearance of intellectual disability is correlated with the amount and position of genetic material lost from chromosome 11. Cognitive testing must be performed carefully and is more difficult to evaluate in children with vision loss.
-
Subjects were categorized as BDNF haploinsufficient by comparative genomic hybridization. Subject A has a large deletion on chromosome 11 that removes one copy of the BDNF gene. Subject B has a smaller deletion that does not remove BDNF.
-
Aniridia. Note the almost complete absence of the iris.





